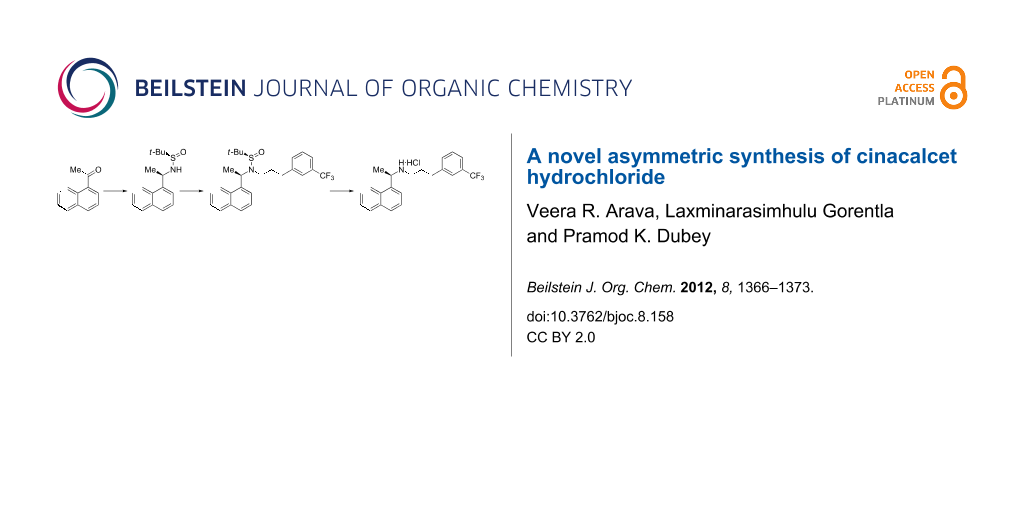
Abstract
A novel route to asymmetric synthesis of cinacalcet hydrochloride by the application of (R)-tert-butanesulfinamide and regioselective N-alkylation of the naphthyl ethyl sulfinamide intermediate is described.

Graphical Abstract
Introduction
Cinacalcet hydrochloride (CNC·HCl, 1, Figure 1) is the first active pharmaceutical ingredient (API) approved by the USFDA for the treatment of secondary hyperparathyroidism. It is sold under the trade names of Sensipar® in USA and Mimpara® in Europe. Hyperparathyroidism (HPT) is a condition characterized by the over-secretion of parathyroid hormone (PTH), a result of the failure of calcium receptors on parathyroid glands [1,2]. Calcimimetics are the agents that mimic the action of calcium to increase the sensitivity of these receptors to calcium, which inhibits the release of parathyroid hormone and lowers PTH levels in a very short time [3]. CNC·HCl (1) is the first and most successful drug among the calcimimetic agents, administered to patients with chronic kidney diseases.
![[1860-5397-8-158-1]](/bjoc/content/figures/1860-5397-8-158-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Cinalcet hydrochloride (CNC·HCl, 1).
Figure 1: Cinalcet hydrochloride (CNC·HCl, 1).
Results and Discussion
Several synthetic approaches have been reported for the synthesis of enantiopure CNC·HCl (1) [4-20]. In our quest to utilize the chiral tert-butanesulfinamides in asymmetric syntheses of chiral amine APIs in an industrial setting [21-23], we report a novel asymmetric synthesis of 1 (Scheme 1) based on (R)-tert-butanesulfinamide (2), which was developed and extensively studied by Ellman [24]. We have chosen 1-acetylnaphthalene (3) (Scheme 1) as a key starting material to produce the chiral amine center, and 3-trifluoromethylbenzaldehyde (8) as another key starting material for the preparation of the intermediates 1-(3-bromopropyl)-3-trifluoromethylbenzene (5) and 1-(3-iodopropyl)-3-trifluoromethylbenzene (6, Scheme 2).
![[1860-5397-8-158-i1]](/bjoc/content/inline/1860-5397-8-158-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-8-158-i2]](/bjoc/content/inline/1860-5397-8-158-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the intermediates 5 and 6 from 8.
Scheme 2: Synthesis of the intermediates 5 and 6 from 8.
First, the enantiopure 2-methylpropane-2-sulfinic acid 1-(naphthalen-1-yl)ethylamide (4) was prepared by the condensation of 2 with 3 according to the earlier reported procedure [23]. The obtained crude is a diastereomeric mixture of 4a and 4b with a ratio of 73:27 (chiral HPLC analysis). From this crude mixture, 4a was isolated in pure form by recrystallization from 10% ethyl acetate–hexanes in 68% yield with 99.94% ee (Scheme 3).
![[1860-5397-8-158-i3]](/bjoc/content/inline/1860-5397-8-158-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Asymmetric synthesis of naphthylethylsulfinamide 4.
Scheme 3: Asymmetric synthesis of naphthylethylsulfinamide 4.
Malonic acid (13) was condensed with 8 and a catalytic amount of piperidine in pyridine under reflux to yield 3-(3-trifluoromethylphenyl)acrylic acid (9) in 90% yield [13]. The acid 9 was hydrogenated in the presence of 10% Pd/C catalyst in aqueous sodium hydroxide solution at ambient temperature, initially under 4.0 bar hydrogen overpressure to get 3-(3-trifluoromethylphenyl)propionic acid (10) in 98% yield. The desfluoro impurity 14 (around 8.2%, HPLC) was also detected (Scheme 4). Initially, 14 was reported in the literature [5] as a carryover impurity from bromo derivative 5 and from hydrogenation of 9 in methanol. The latter also lead to the reduction of the phenyl ring concomitant with double bond reduction. To reduce the formation of 14, the reduction was tried to be carried out with 5% Pd/C and simply bubbling hydrogen gas into the reaction mixture at room temperature, and also by reducing the proportion of the catalyst (Table 1). The impurity formation was observed even at low catalyst loading. As a result, the impurity was removed by recrystallizing the crude product (Table 1, entry 5) in n-hexane at 0 °C twice and the pure product was obtained in 90% yield. The obtained product purity was >99% (GC–MS) which was sufficient for further conversions.
![[1860-5397-8-158-i4]](/bjoc/content/inline/1860-5397-8-158-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Table 1: Screening of reduction conditions (9→10).
| entry | catalyst | H2 overpressure (bar) | time (h) | temperature (°C) | crude yield (%)a | 10 (%) | 14 (%) | |
|---|---|---|---|---|---|---|---|---|
| Pd/C (%) | (mol %) | |||||||
| 1 | 10 | 10 | 4.0 | 1 | 30–35 | 98 | 91.6b | 8.2b |
| 2 | 5 | 10 | 4.0 | 3 | 30–35 | 95 | 97.7b | 2.2b |
| 3 | 5 | 10 | 0c | 5 | 30–35 | 96 | 87.3d | 12.6d |
| 4 | 5 | 5 | 0c | 5 | 25–30 | 94 | 94.9d | 5.1d |
| 5 | 5 | 2 | 0c | 7 | 25–30 | 95 | 98.8d | 1.2d |
aIsolated yield; b% yield from HPLC; cbubbling (1 atm); d% yield in GC–MS.
We determined that 14 was not a carryover impurity from 8 by spiking analysis of 8 and 15 in gas chromatography. Both were clearly separated and 15 was absent in 8. The impurity 14 was further structurally assigned by its synthetic preparation starting from 15.
The obtained propionic acid 10 was esterified to its methyl ester 11 with methanol and thionyl chloride under reflux for 4 h in 97% yield [13]. This ester 11 was reduced with NaBH4 in THF and methanol under reflux for 5 h [25] to afford 3-(3-trifluoromethylphenyl)propan-1-ol (12) in 95% yield (Scheme 5).
![[1860-5397-8-158-i5]](/bjoc/content/inline/1860-5397-8-158-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of alcohol intermediate 12 from 10.
Scheme 5: Synthesis of alcohol intermediate 12 from 10.
From this alcohol 12, both the bromo 5 and iodo 6 derivatives were prepared. Bromo derivative 5 was prepared simply by heating 12 in 48% aqueous HBr solution under reflux for 15 h. The obtained crude was purified by passing it through a silica gel plug with n-hexane to afford 5 in 82% yield. Iodide 6 was prepared by reacting 12 with molecular iodine in the presence of triphenylphosphine and imidazole in CH2Cl2 at room temperature for 2 h to give the product in 85% yield (Scheme 6).
![[1860-5397-8-158-i6]](/bjoc/content/inline/1860-5397-8-158-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of bromo 5 and iodo 6 derivatives.
Scheme 6: Synthesis of bromo 5 and iodo 6 derivatives.
Regioselective N-alkylation of N-tert-butanesulfinamides was difficult to achieve as S-alkylation can also be possible [26]. To our knowledge, limited procedures were reported for the regioselective N-alkylation of N-tert-butanesulfinylamides [27,28]. Following the procedure reported in literature [26], initial experiments were conducted with 4 and 5, and 4 and 6 in DMF with 2.0 equiv of LiHMDS at −20 °C to room temperature, which resulted in an isolated yield of 40 and 44% pure product, respectively. To force the reaction to completion and to improve the yield, the alkylation was carried out in various combinations and under various conditions (Table 2). Surprisingly, reactions without DMF and only in solvents like THF or DMSO were not initiated at all. Different combinations of bases and catalysts were also screened for the reaction progress, i.e., n-BuLi, LDA, Cs2CO3 with dppf/PdCl2, K2CO3 with Cu(I)/L-proline, NaH and TEA. All of them failed to initiate the reaction. Only LiHMDS worked well for the regioselective N-alkylation of sulfinylamide 4a in a better yield (72% isolated pure product) than the earlier reported procedures for 1 (Scheme 7). Finally, hydrolysis of 7 dissolved in MTBE with conc. HCl at ambient temperature liberated the pure 1 (Scheme 8).
Table 2: Conditions for regioselective N-alkylation of naphthylethylsulfinamide 4a.
| entry | intermediate | base/solvent/catalyst | time (h) | temperature (°C) | 7 yield (%)a |
|---|---|---|---|---|---|
| 1 | 5 | LiHMDS (2.0 equiv)/DMF:THF | 6 | −20 to rt | 41 |
| 2 | 5 | LiHMDS (2.5 equiv)/DMF:THF | 6 | −20 to rt | 44 |
| 3 | 5 | LiHMDS (3.0 equiv)/DMF:THF | 6 | −20 to rt | 50 |
| 4 | 5 | LiHMDS (4.0 equiv)/DMF:THF | 6 | −20 to rt | 66 |
| 5 | 5 | LiHMDS (5.0 equiv)/DMF:THF | 6 | −20 to rt | 69 |
| 6 | 5 | LiHMDS (7.0 equiv)/DMF:THF | 6 | −20 to rt | 70 |
| 7 | 5 | LiHMDS (2.0 equiv)/THF | 24 | −20 to rt | NR |
| 8 | 5 | KOt-Bu (2.0 equiv)/DMF:THF | 24 | −20 to rt | 10 |
| 9 | 6 | LiHMDS (2.0 equiv)/DMF:THF | 6 | −20 to rt | 44 |
| 10 | 6 | LiHMDS (2.5 equiv)/DMF:THF | 6 | −20 to rt | 48 |
| 11 | 6 | LiHMDS (3.0 equiv)/DMF:THF | 6 | rt | 55 |
| 12 | 6 | LiHMDS (4.0 equiv)/DMF:THF | 6 | rt | 70 |
| 13 | 6 | LiHMDS (5.0 equiv)/DMF:THF | 6 | rt | 71 |
| 14 | 6 | LiHMDS (7.0 equiv)/DMF:THF | 6 | rt | 72 |
| 15 | 6 | LiHMDS (2.0 equiv)/THF | 24 | rt | NR |
| 16 | 5 | LiHMDS (4.0 equiv)/THF/Pd(dppf)Cl2 | 6 | −20 to rt | NR |
| 17 | 5 | n-BuLi (1.1 equiv)/DMF:THF | 24 | −20 to rt | NR |
| 18 | 5 | LDA (2.0 equiv)/DMF:THF | 24 | −20 to rt | NR |
| 19 | 5 | Cs2CO3 (2.5 equiv)/DMSO/Pd(dppf)sCl2 | 24 | rt | NR |
| 20 | 5 | K2CO3 (2.0 equiv)/NMP/CuI:L-proline | 24 | rt | NR |
| 21 | 5 | NaH (1.5 equiv)/THF | 24 | 0 to 65 | NR |
| 22 | 5 | TEA (2.0 equiv)/THF | 24 | rt | NR |
| 23 | 12 | THF/Raney-Ni | 24 | rt | NR |
aIsolated yield (NR: no reaction).
![[1860-5397-8-158-i7]](/bjoc/content/inline/1860-5397-8-158-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Regioselective N-alkylation of naphthyl ethyl sulfinamide 4a.
Scheme 7: Regioselective N-alkylation of naphthyl ethyl sulfinamide 4a.
![[1860-5397-8-158-i8]](/bjoc/content/inline/1860-5397-8-158-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Acid hydrolysis of N-tert-butanesulfinyl group in 7.
Scheme 8: Acid hydrolysis of N-tert-butanesulfinyl group in 7.
Conclusion
In summary, a novel stereoselective and short synthesis of (R)-cinacalcet hydrochloride by the application of (R)-tert-butanesulfinamide and regioselective N-alkylation of the naphthylethylsulfinamide intermediate was achieved in good yield.
Experimental
Experiments were conducted under nitrogen atmosphere unless stated otherwise. All solvents and reagents were reagent grade pure and used without further purification. All melting points were determined on Polmon MP-96 melting point apparatus. 1H and 13C NMR spectra were recorded using a Bruker 400 MHz spectrometer (400 and 100 MHz, respectively) with TMS as internal standard. Mass spectra were recorded on a Perkin-Elmer mass spectrometer operating at an ionization potential of 70 eV. IR spectra were recorded on a Perkin-Elmer spectrophotometer as KBr pellets or neat. Analytical TLC is conducted on E-Merck 60 F254 aluminium-packed silica gel plates (0.2 mm). Developed plates were visualized under UV light or in an iodine chamber. Chiral HPLC analyses were recorded with on a Waters Alliance 2695 chromatograph with a 2487 UV detector.
Preparation of 2-methylpropane-2-sulfinic acid (1-naphthalen-1-ylethyl)amide (4): (R)-tert-Butanesulfinamide (2) (16.14 g, 0.133 mol) was added to a solution of titanium tetraethoxide (54.0 g, 0.236 mol) and 1-acetylnaphthalene (3, 20.0 g, 0.117 mol) in THF (200.0 mL) under an N2 atmosphere and the mixture was refluxed at 65–70 °C for 30 h. Upon completion, as determined by TLC, the reaction mixture was first cooled to rt and then to −48 to −52°C with a dry ice/acetone bath. NaBH4 (6.66 g, 0.176 mol) was added portion wise at −48 to −52 °C, and the mixture was stirred at −48 °C until the reduction was complete. Then, methanol (20.0 mL) was added drop wise until gas evolution stopped. The resulting mixture was poured into an equal volume of brine with rapid stirring. The resulting suspension was filtered through a pad of celite, and the bed was washed with ethyl acetate. The filtrate was extracted with ethyl acetate. The combined organic portions were dried over anhydrous Na2SO4, filtered, and concentrated to obtain the crude product. This crude product was crystallized from an n-hexane/ethyl acetate mixture (9:1 ratio) to get pure 4 as a pale yellow crystalline solid. The remaining product from the mother liquor was isolated from column chromatography with n-hexane/ethyl acetate (9:1). Yield = 22.0 g (68%); mp 97.0–98.2 °C; [α]D23 −116.8 (c 1.0, CHCl3); chiral purity (HPLC): 99.97% (R-isomer) and 0.03% (S-isomer); IR (KBr): 3220 (sharp, strong, –NH), 1057 (sharp, strong, –SO) cm−1; 1H NMR (CDCl3/TMS) δ 1.24 (s, 9H, C(CH3)3), 1.70 (d, J = 6.5, 3H, –CH3), 3.62 (s, 1H, –NH, D2O exchangeable), 5.39 (q, 1H, –CH), 7.46–7.62 (m, 4H, ArH), 7.81 (dd, J1 = 29.0, J2 = 8.0, 2H, ArH), 8.24 (d, J = 8.4, 1H, ArH); 13C NMR (CDCl3/TMS) δ 139.05, 133.88, 130.36, 128.89, 128.35, 126.40, 125.72, 123.42, 123.01, 55.40, 49.21, 22.55, 21.71; MS m/z: 276 [M + 1]+.
Preparation of 2-methylpropane-2-sulfinic acid (1-naphthalen-1-ylethyl)-[3-(3-trifluoromethylphenyl)propyl]amide (7): Amide 4 (3.0 g, 0.010 mol) was dissolved in DMF (9.0 mL) at rt under an N2 atmosphere. To the resulting solution, LiHMDS (63.85 mL, 0.070 mol) was added drop wise at rt over a period of 1 h. After stirring for 10 min, 6 (3.14 g, 0.010 mol) diluted with THF (3.0 mL) was added at rt over a period of 15 min, stirred for another 6 h at rt, and 15% ammonium chloride solution (30.0 mL) was added slowly. Then, the product was extracted with ethyl acetate. Evaporation of solvent and column chromatography of the crude product furnished 7 as a thick pale yellow syrup. Yield = 3.61 g (72%); chiral purity (HPLC): 99.9%; IR (neat): absence of 3220 (–NH), 2949, 2868, 1327, 1162, 1124, 1072, 798 and 779 cm−1; 1H NMR (CDCl3/TMS) δ 0.80 (s, 9H, C(CH3)3), 1.78 (d, J = 6.7, 3H, –CH3), 1.98 (m, 1H, –CH2), 2.28 (m, 1H, –CH2), 2.66 (m, 2H, –CH2), 2.90 (m, 1H, –CH2), 3.31 (m, 1H, –CH2), 5.29 (q, 1H, –CH), 7.22–7.53 (m, 6H, ArH), 7.60 (d, J = 7.0, 1H, ArH), 7.79 (d, J = 8.1, 1H, ArH), 7.86 (d, J = 7.7, 1H, ArH), 8.12 (d, J = 8.4, 1H, ArH); 13C NMR (CDCl3/TMS) δ 142.0, 137.16, 133.72, 131.75, 131.24, 130.81, 128.88, 128.77, 128.28, 126.10, 125.44, 125.22, 124.82, 123.02, 122.84, 57.27, 54.26, 43.13, 33.35, 30.84, 23.10, 18.47; MS m/z: 462 [M + 1]+.
Preparation of 3-(3-trifluoromethylphenyl)acrylic acid (9): Aldehyde 8 (50.0 g, 0.287 mol) was dissolved in pyridine (100.0 mL) and piperidine (0.5 mL) at rt. Malonic acid (13, 80.0 g, 0.768 mol) was added to this solution at rt and it was stirred for 15 min. Then, the reaction mixture was heated to 115–120 °C and stirred for another 4 h. After completion of the reaction, the reaction mixture was cooled to rt, water (500.0 mL) was added and its pH was adjusted to 2 with conc. HCl solution (50.0 mL). The precipitated white solid was filtered and washed with water. The solid was dried at 70–75 °C to a constant weight to give 9. Yield = 56.0 g (90%); purity (HPLC): 99.9%; IR (KBr): 3220 (–COOH), 1682 (–CO–) cm−1; 1H NMR (CDCl3/TMS) δ 6.50 (d, 1H, –CH), 7.54 (t, 1H, ArH), 7.63 (d, 2H, –CH and ArH), 7.68–7.75 (m, 2H, ArH); 13C NMR (CDCl3/TMS) δ 172.62, 147.10, 140.03, 135.72, 134.15, 131.0, 129.80, 129.07, 129.03, 127.09, 125.71.
Preparation of 3-(3-trifluoromethylphenyl)propionic acid (10): Acid 9 (50.0 g, 0.231 mol) was dissolved in aqueous NaOH solution (500.0 mL) at rt. To the resulting clear solution, 5% Pd/C (1.0 g) was added and it was hydrogenated by bubbling H2 gas (1 bar) at 25–30 °C for 7 h. The reaction mixture was filtered through a pad of celite to separate the Pd/C catalyst. The pH of the filtrate layer was adjusted to 2 with conc. HCl solution (35.0 mL) at rt and the product was extracted with ethyl acetate (2 × 250.0 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated under vacuum to get the product as colorless oil, which was recrystallized twice from n-hexane at 0 °C. Decanting the hexane layer from the crystalline solid affords the pure 10 (clear liquid at 25 °C). Yield = 47.3 g (90%); purity (GC–MS) = 99.45%; IR (neat): 3300–3400 (–COOH), 1712 (–CO–) cm−1; 1H NMR (CDCl3/TMS) δ 2.72 (t, 2H, –CH2), 3.03 (t, 2H, –CH2), 7.41 (d, 2H, ArH), 7.48 (s, 2H, ArH), 10.5 (broad s, 1H, –COOH); 13C NMR (CDCl3/TMS) δ 178.69, 140.92, 131.59, 130.97 (q), 128.89, 124.93, 123.9 (q), 123.23, 35.11, 30.17.
Preparation of 3-(3-trifluoromethylphenyl)propionic acid methyl ester (11): Acid 10 (50.0 g, 0.229 mol) was dissolved in methanol (200.0 mL) at rt and thionyl chloride (27.28 g, 0.229 mol) was added to this solution at rt. The clear solution was heated under refluxfor 4 h. After completion of the reaction, methanol was distilled off and the remaining reaction mixture was cooled to rt. Then, water (200.0 mL) was added and the pH of the mixture was adjusted to 7 with 5% aqueous sodium bicarbonate solution (150.0 mL). The product was extracted with ethyl acetate (2 × 250.0 mL) and the organic layer was dried over anhydrous Na2SO4. Then it was evaporated under vacuum to get the product as colorless oil. Yield = 52.0 g (97%); purity (GC) = 99.5%; IR (neat): absence of 3300–3400 (–COOH), 1740 (–CO–) cm−1; 1H NMR (CDCl3/TMS) δ 2.65 (t, J = 7.6, 2H, –CH2), 3.01 (t, J = 7.6, 2H, –CH2), 3.67 (s, 3H, –OCH3), 7.39 (d, J = 8.8, 2H, ArH), 7.46 (s, 2H, ArH); 13C NMR (CDCl3/TMS) δ 172.70, 141.31, 131.62, 130.72 (q), 128.80, 124.90, 123.99 (q), 123.09, 51.50, 35.17, 30.53.
Preparation of 3-(3-trifluoromethylphenyl)propan-1-ol (12): Compound 11 (50.0 g, 0.215 mol) was dissolved in THF (500.0 mL) and sodium borohydride (49.0 g, 1.295 mol) was added at rt. The resulting reaction solution was heated under reflux for 10 min. Methanol (500.0 mL) was added to this solution at 60–65 °C over a period of 4 h. After complete addition of methanol, the reaction solution was stirred for another 5 h to complete the reduction. After completion of the reaction, the solvent was distilled off and the remaining reaction mixture was cooled to rt, water (500.0 mL) was added and the pH was adjusted to 5 with conc. HCl solution (50.0 mL). The product was extracted with ethyl acetate (2 × 250.0 mL) and the organic layer was dried over anhydrous sodium sulfate and evaporated under vacuum to get the product as colorless oil. Yield = 43.5 g (95%); purity (GC) = 99.7%; IR (neat) 3350 (–OH), 2941, 2870, 1450, 1331, 1162, 1124, 1073 cm−1; 1H NMR (CDCl3/TMS) δ 1.90 (m, 2H, –CH2), 2.08 (s, 1H, –OH), 2.77 (t, J = 7.8, 2H, –CH2), 3.67 (t, J = 6.3, 2H, –CH2), 7.38 (d, J = 5.3, 2H, ArH), 7.45 (d, J = 5.3, 2H, ArH); 13C NMR (CDCl3/TMS) δ 142.64, 131.75, 130.54 (q), 128.67, 125.50, 124.99, 124.96, 122.8, 122.66, 122.63, 61.69, 33.80, 31.71.
Preparation of 1-(3-bromopropyl)-3-trifluoromethylbenzene (5): Compound 12 (50.0 g, 0.244 mol) was added to 48% aqueous HBr solution (400.0 mL) at rt. The reaction mixture was heated to 85–90 °C and stirred for 15 h. After completion of the reaction, the mixture was cooled to rt and water (250.0 mL) was added. The product was extracted with n-hexane (2 × 250.0 mL) and the organic layer was dried over anhydrous sodium sulfate and evaporated under vacuum to get the crude product, which was passed through a silica gel plug in n-hexane to afford the pure product as colorless oil. Yield = 53.6 g (82%); purity (GC) = 99.0%; IR (neat): absence of 3350 (–OH), 1451, 1330, 1164, 1125, 1094, 799, 702 cm−1; 1H NMR (CDCl3/TMS) δ 2.19 (quintet, 2H, –CH2), 2.86 (t, J = 7.3, 2H, –CH2), 3.41 (t, J = 6.5, 2H, –CH2) and 7.39–7.49 (m, 4H, ArH); 13C NMR (CDCl3/TMS) δ 141.36, 131.87, 130.67 (q, CF3), 128.83, 125.07, 123.03, 33.70, 33.67, 32.52.
Preparation of 1-(3-iodopropyl)-3-trifluoromethylbenzene (6): Compound 12 (50.0 g, 0.244 mol) was dissolved in CH2Cl2 (150.0 mL) and imidazole (2.0 g, 0.029 mol) and triphenylphosphine (70.64 g, 0.269 mol) were added at rt. To the resulting pale yellow clear solution was added iodine (62.0 g, 0.244 mol) in portions at less than 35 °C over a period of 1 h. The reaction mixture was stirred for 1 h and quenched with saturated sodium thiosulfate solution (2 × 100.0 mL) and water (250.0 mL). The product was extracted with n-hexane (2 × 250.0 mL) and the organic layer was dried over anhydrous sodium sulfate and evaporated under vacuum to get the product as colorless oil. Yield = 65.3 g (85%); purity (GC) = 99.45%; IR (neat): absence of 3100–3400 (–OH), 1450, 1330, 1164, 1126, 1074, 797, 702 cm−1; 1H NMR (CDCl3/TMS) δ 2.14 (quintet, 2H, –CH2), 2.80 (t, J = 7.3, 2H, –CH2), 3.41 (t, J = 6.6, 2H, –CH2), 7.39–7.49 (m, 4H, ArH); 13C NMR (CDCl3/TMS) δ 141.34, 132.01, 131.02 (q, CF3), 128.97, 125.53, 123.26, 36.02, 34.55, 5.72.
Preparation of cinacalcet hydrochloride (1): Compound 7 (3.0 g, 0.0065 mol) and MTBE (15.0 mL) were stirred for 15 min to become a clear solution. Conc. HCl solution (3.2 mL, 0.0129 mol) was added drop wise and stirred for 15 min at rt. The material was filtered, washed with MTBE and recrystallized from acetonitrile and water (1:2 ratio) at 65 °C for 2 h. The solid was dried at 50–55 °C to a constant weight to give pure hydrochloride salt of 1. Yield = 2.1 g (91%); chiral purity (HPLC) = 99.95%; mp 174.6–176.8 °C; IR (KBr): 3427 (broad, –NH–), 2951, 2797, 2750, 2712, 1587, 1450, 1327, 1165, 1128, 1072, 798, 775 cm−1; 1H NMR (DMSO-d6/TMS) δ 1.67 (d, J = 6.6, 3H, –CH3), 1.99 (quintet, 2H, –CH2), 2.70 (m, 2H, –CH2), 2.93 (m, 2H, –CH2), 5.30 (q, 1H, –CH), 7.46–7.61 (m, 7H, ArH), 7.95–8.03 (m, 3H, ArH), 8.23 (d, J = 8.0, 1H, ArH), 9.36 (s, 1H, –NH) and 10.04 (s, 1H, HCl); 13C NMR (DMSO-d6/TMS) δ 142.61, 134.48, 133.70, 132.80, 130.64, 129.74, 129.58,129.28, 127.29, 126.54, 125.90, 125.08, 124.67, 123.16, 122.98, 52.37, 45.04, 31.84, 27.44, 20.30; MS m/z: 358 [M + 1]+.
Supporting Information
| Supporting Information File 1: 1H NMR, 13C NMR and ESI–MS spectra of compounds 1, 4, 5, 6 and 7. | ||
| Format: PDF | Size: 674.0 KB | Download |
References
-
Sorbera, L. A.; Castaner, R. M.; Bayes, M. Drugs Future 2002, 27, 831–836. doi:10.1358/dof.2002.027.09.695016
Return to citation in text: [1] -
Franceschini, N.; Joy, M. S.; Kshirsagar, A. Expert Opin. Invest. Drugs 2003, 12, 1413–1421. doi:10.1517/13543784.12.8.1413
Return to citation in text: [1] -
Hebert, S. C. Annu. Rev. Med. 2006, 57, 349–364. doi:10.1146/annurev.med.57.121304.131328
Return to citation in text: [1] -
Reddy, B. P.; Reddy, K. R.; Reddy, D. M.; Reddy, R. R.; Reddy, J. M.; Krishna, B. V. Process for cinacalcet hydrochloride. WO2012007954, Jan 19, 2012.
Return to citation in text: [1] -
Shinde, G. B.; Niphade, N. C.; Deshmukh, S. P.; Toche, R. B.; Mathad, V. T. Org. Process Res. Dev. 2011, 15, 455–461. doi:10.1021/op200016a
Return to citation in text: [1] [2] -
Velez, M. B.; Mangion, B.; Repasi, J.; Szabo, A.; Szekeres, T. Process for preparing cinacalcet hydrochloride and polymorphic forms thereof. US020110295037A1, Dec 1, 2011.
Return to citation in text: [1] -
Sebastian, S.; Sarma, S. R.; Ramamurthy, K.; Pradhan, N. S. Unsaturated cinacalcet salts and process for preparing cinacalcet hydrochloride. US020110178326A1, July 21, 2011.
Return to citation in text: [1] -
Chidambaram, V.; Prajapaty, R.; Johar, P. S.; Sharma, E.; Wadhwa, L. Process for preparing cinacalcet and pharmaceutically acceptable salts thereof. US020110172455A1, July 14, 2011.
Return to citation in text: [1] -
Ferrari, M.; Ghezzi, M.; Bonaldi, M. Process for the synthesis of cinacalcet hydrochloride. US020110105799A1, May 5, 2011.
Return to citation in text: [1] -
Allegrini, P.; Attolino, E.; Rossi, D. Process for the preparation of cinacalcet and intermediates thereof. US020110124917A1, May 26, 2011.
Return to citation in text: [1] -
Sebastian, S.; Sarma, S. R.; Ramamurthy, K.; Pradhan, N. S. Highly pure cinacalcet or a pharmaceutically acceptable salt thereof. WO02010067204A1, July 17, 2010.
Return to citation in text: [1] -
Thiel, O.; Bernard, C.; Larsen, R.; Martinelli, J. M.; Raza, M. T. Methods of synthesizing cinacalcet and salts thereof. WO2009002427A3, Dec 31, 2008.
Return to citation in text: [1] -
Bijukumar, G.; Maloyesh, B.; Bhaskar, B. S.; Rajendra, A. Synth. Commun. 2008, 38, 1512–1517. doi:10.1080/00397910801928541
Return to citation in text: [1] [2] [3] -
Thiel, O. R.; Bernard, C.; Tormos, W.; Brewin, A.; Hirotani, S.; Murakami, K.; Saito, K.; Larsen, R. D.; Martinelli, M. J.; Reider, P. J. Tetrahedron Lett. 2008, 49, 13–15. doi:10.1016/j.tetlet.2007.11.030
Return to citation in text: [1] -
Reddy, P. P.; Akundi, S. P.; Suthrapu, S. K.; Kolla, N. K.; Kotagiri, V. K.; Neelam, U. K.; Baddam, S. R.; Manne, N. Process for the preparation of cinacalcet. WO02008058235, May 15, 2008.
Return to citation in text: [1] -
Allegrini, P.; Attolino, E. Process for the preparation of cinacalcet. US20080319229A1, Dec 25, 2008.
Return to citation in text: [1] -
Lifshitz-Liron, R. Process for the preparation of cinacalcet base. US000007393967B2, Jan 7, 2008.
Return to citation in text: [1] -
Avhar-Maydan, S.; Eisenstadt, A.; Lifshitz-Liron, R.; Raizi, Y.; Ramaty, R.; Teva, P.; Wizel, S. Process for preparing cinacalcet hydrochloride. WO2006125026A3, Nov 23, 2006.
Return to citation in text: [1] -
Balandrin, M. F.; Delmar, E. G.; Moe, S. T.; Nemeth, E. F.; van Wagenen, B. C. Calcium receptor-active compounds. US000006211244B1, April 3, 2001.
Return to citation in text: [1] -
Balandrin, M. F.; Delmar, E. G.; Moe, S. T.; Nemeth, E. F.; van Wagenen, B. C. Calcium receptor-active molecules US000006011068A, Jan 4, 2000.
Return to citation in text: [1] -
Reddy, A. V.; Rao, S. U. B.; Narasimha, G. L.; Dubey, P. K. Synth. Commun. 2009, 39, 1451–1456. doi:10.1080/00397910802519216
Return to citation in text: [1] -
Arava, V. R.; Laxminarasimhulu, G.; Dubey, P. K. Der Pharma Chemica 2011, 3, 426–433.
Return to citation in text: [1] -
Arava, V. R.; Gorentla, L.; Dubey, P. K. Int. J. Org. Chem 2011, 1, 26–32. doi:10.4236/ijoc.2011.12005
Return to citation in text: [1] [2] -
Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600–3740. doi:10.1021/cr900382t
Return to citation in text: [1] -
Saeed, A.; Ashraf, Z. J. Chem. Sci. 2006, 118, 419–423. doi:10.1007/BF02711452
Return to citation in text: [1] -
Martjuga, M.; Belyakov, S.; Liepinsh, E.; Suna, E. J. Org. Chem. 2011, 76, 2635–2647. doi:10.1021/jo1025767
Return to citation in text: [1] [2] -
Ruano, J. L. G.; Parra, A.; Alemán, J.; Yuste, F.; Mastranzo, V. M. Chem. Commun. 2009, 45, 404–406. doi:10.1039/b816846f
Return to citation in text: [1] -
Cano, R.; Ramón, D. J.; Yus, M. J. Org. Chem. 2011, 76, 5547–5557. doi:10.1021/jo200559h
Return to citation in text: [1]
| 1. | Sorbera, L. A.; Castaner, R. M.; Bayes, M. Drugs Future 2002, 27, 831–836. doi:10.1358/dof.2002.027.09.695016 |
| 2. | Franceschini, N.; Joy, M. S.; Kshirsagar, A. Expert Opin. Invest. Drugs 2003, 12, 1413–1421. doi:10.1517/13543784.12.8.1413 |
| 24. | Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600–3740. doi:10.1021/cr900382t |
| 21. | Reddy, A. V.; Rao, S. U. B.; Narasimha, G. L.; Dubey, P. K. Synth. Commun. 2009, 39, 1451–1456. doi:10.1080/00397910802519216 |
| 22. | Arava, V. R.; Laxminarasimhulu, G.; Dubey, P. K. Der Pharma Chemica 2011, 3, 426–433. |
| 23. | Arava, V. R.; Gorentla, L.; Dubey, P. K. Int. J. Org. Chem 2011, 1, 26–32. doi:10.4236/ijoc.2011.12005 |
| 4. | Reddy, B. P.; Reddy, K. R.; Reddy, D. M.; Reddy, R. R.; Reddy, J. M.; Krishna, B. V. Process for cinacalcet hydrochloride. WO2012007954, Jan 19, 2012. |
| 5. | Shinde, G. B.; Niphade, N. C.; Deshmukh, S. P.; Toche, R. B.; Mathad, V. T. Org. Process Res. Dev. 2011, 15, 455–461. doi:10.1021/op200016a |
| 6. | Velez, M. B.; Mangion, B.; Repasi, J.; Szabo, A.; Szekeres, T. Process for preparing cinacalcet hydrochloride and polymorphic forms thereof. US020110295037A1, Dec 1, 2011. |
| 7. | Sebastian, S.; Sarma, S. R.; Ramamurthy, K.; Pradhan, N. S. Unsaturated cinacalcet salts and process for preparing cinacalcet hydrochloride. US020110178326A1, July 21, 2011. |
| 8. | Chidambaram, V.; Prajapaty, R.; Johar, P. S.; Sharma, E.; Wadhwa, L. Process for preparing cinacalcet and pharmaceutically acceptable salts thereof. US020110172455A1, July 14, 2011. |
| 9. | Ferrari, M.; Ghezzi, M.; Bonaldi, M. Process for the synthesis of cinacalcet hydrochloride. US020110105799A1, May 5, 2011. |
| 10. | Allegrini, P.; Attolino, E.; Rossi, D. Process for the preparation of cinacalcet and intermediates thereof. US020110124917A1, May 26, 2011. |
| 11. | Sebastian, S.; Sarma, S. R.; Ramamurthy, K.; Pradhan, N. S. Highly pure cinacalcet or a pharmaceutically acceptable salt thereof. WO02010067204A1, July 17, 2010. |
| 12. | Thiel, O.; Bernard, C.; Larsen, R.; Martinelli, J. M.; Raza, M. T. Methods of synthesizing cinacalcet and salts thereof. WO2009002427A3, Dec 31, 2008. |
| 13. | Bijukumar, G.; Maloyesh, B.; Bhaskar, B. S.; Rajendra, A. Synth. Commun. 2008, 38, 1512–1517. doi:10.1080/00397910801928541 |
| 14. | Thiel, O. R.; Bernard, C.; Tormos, W.; Brewin, A.; Hirotani, S.; Murakami, K.; Saito, K.; Larsen, R. D.; Martinelli, M. J.; Reider, P. J. Tetrahedron Lett. 2008, 49, 13–15. doi:10.1016/j.tetlet.2007.11.030 |
| 15. | Reddy, P. P.; Akundi, S. P.; Suthrapu, S. K.; Kolla, N. K.; Kotagiri, V. K.; Neelam, U. K.; Baddam, S. R.; Manne, N. Process for the preparation of cinacalcet. WO02008058235, May 15, 2008. |
| 16. | Allegrini, P.; Attolino, E. Process for the preparation of cinacalcet. US20080319229A1, Dec 25, 2008. |
| 17. | Lifshitz-Liron, R. Process for the preparation of cinacalcet base. US000007393967B2, Jan 7, 2008. |
| 18. | Avhar-Maydan, S.; Eisenstadt, A.; Lifshitz-Liron, R.; Raizi, Y.; Ramaty, R.; Teva, P.; Wizel, S. Process for preparing cinacalcet hydrochloride. WO2006125026A3, Nov 23, 2006. |
| 19. | Balandrin, M. F.; Delmar, E. G.; Moe, S. T.; Nemeth, E. F.; van Wagenen, B. C. Calcium receptor-active compounds. US000006211244B1, April 3, 2001. |
| 20. | Balandrin, M. F.; Delmar, E. G.; Moe, S. T.; Nemeth, E. F.; van Wagenen, B. C. Calcium receptor-active molecules US000006011068A, Jan 4, 2000. |
| 26. | Martjuga, M.; Belyakov, S.; Liepinsh, E.; Suna, E. J. Org. Chem. 2011, 76, 2635–2647. doi:10.1021/jo1025767 |
| 3. | Hebert, S. C. Annu. Rev. Med. 2006, 57, 349–364. doi:10.1146/annurev.med.57.121304.131328 |
| 13. | Bijukumar, G.; Maloyesh, B.; Bhaskar, B. S.; Rajendra, A. Synth. Commun. 2008, 38, 1512–1517. doi:10.1080/00397910801928541 |
| 26. | Martjuga, M.; Belyakov, S.; Liepinsh, E.; Suna, E. J. Org. Chem. 2011, 76, 2635–2647. doi:10.1021/jo1025767 |
| 5. | Shinde, G. B.; Niphade, N. C.; Deshmukh, S. P.; Toche, R. B.; Mathad, V. T. Org. Process Res. Dev. 2011, 15, 455–461. doi:10.1021/op200016a |
| 27. | Ruano, J. L. G.; Parra, A.; Alemán, J.; Yuste, F.; Mastranzo, V. M. Chem. Commun. 2009, 45, 404–406. doi:10.1039/b816846f |
| 28. | Cano, R.; Ramón, D. J.; Yus, M. J. Org. Chem. 2011, 76, 5547–5557. doi:10.1021/jo200559h |
| 13. | Bijukumar, G.; Maloyesh, B.; Bhaskar, B. S.; Rajendra, A. Synth. Commun. 2008, 38, 1512–1517. doi:10.1080/00397910801928541 |
| 23. | Arava, V. R.; Gorentla, L.; Dubey, P. K. Int. J. Org. Chem 2011, 1, 26–32. doi:10.4236/ijoc.2011.12005 |
| 25. | Saeed, A.; Ashraf, Z. J. Chem. Sci. 2006, 118, 419–423. doi:10.1007/BF02711452 |
© 2012 Arava et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)