Abstract
The pH-activated E/Z isomerization of a series of hydrazone-based systems having different functional groups as part of the rotor (R = COMe, CN, Me, H), was studied. The switching efficiency of these systems was compared to that of a hydrazone-based molecular switch (R = COOEt) whose E/Z isomerization is fully reversible. It was found that the nature of the R group is critical for efficient switching to occur; the R group should be a moderate H-bond acceptor in order to (i) provide enough driving force for the rotor to move upon protonation, and (ii) stabilize the obtained Z configuration, to achieve full conversion.

Graphical Abstract
Findings
Nature is full of elegant examples of perfectly designed biological motors and machines [1] that perform delicate and precise tasks. Primitive as they may be, numerous artificial molecular machines [2-6] have been developed that strive to mimic their biological counterparts as far as function is concerned. As part of these efforts, a variety of molecular systems have been developed that can perform different types of motion (e.g., translation, rotation) in response to chemical [7-9], electrochemical [10-13], and photochemical stimuli [14-18]. One of the benefits of artificial molecular switches and machines is that their output can be controlled or fine-tuned by altering their components [19-21]. A relevant example in this context is Feringa’s overcrowded alkene-based light-driven rotary switches that can be induced to rotate at different rates by replacing a naphthyl group in the upper-half of the molecule (i.e., the rotor) with a less sterically hindered benzothiophenyl group [20].
Previously, we have shown that hydrazone-based rotary switches can change their configuration (i.e., E/Z isomerization) as a function of pH [22-24], or upon the addition of a Lewis acid (i.e., Zn2+) [25]. The simplest hydrazone switch (PPH-1, Scheme 1) for example, exists mainly as its E isomer (PPH-1-E) in solution, as illustrated by the E/Z isomer ratio of 93:7 in CD3CN. Protonation of PPH-1-E with acid results in an intermediate PPH-1-E-H+, which quickly isomerizes to PPH-1-Z-H+, which is the more stable isomer. During this process, an E/Z isomerization takes place, which can be fully reversed by the addition of base to the solution.
![[1860-5397-8-98-i1]](/bjoc/content/inline/1860-5397-8-98-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The acid-activated switching process of PPH-1.
Scheme 1: The acid-activated switching process of PPH-1.
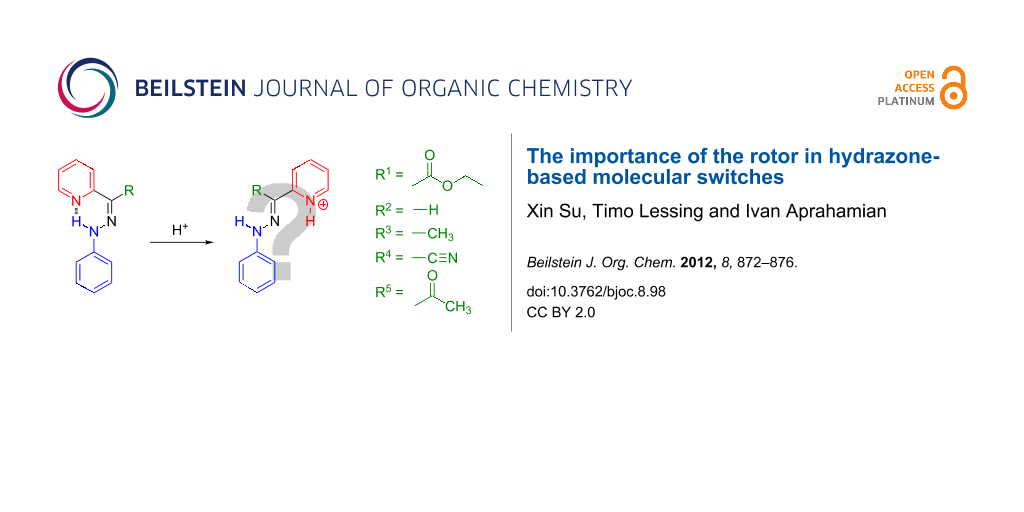
In order to fine tune the properties of the hydrazone switches, we studied the effect of different R groups in the rotor part (Scheme 2) on the switching cycle. The target hydrazones were synthesized either by the direct condensation of phenylhydrazine with the corresponding aldehyde (PPH-2) or ketone (PPH-3), or by Japp–Klingemann reaction (PPH-4, PPH-5) [26]. The NMR spectroscopy and mass spectrometry characterizations show results consistent with the previously reported data [26].
![[1860-5397-8-98-i2]](/bjoc/content/inline/1860-5397-8-98-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The hydrazone-based molecular systems that were analysed in this paper, each having different rotors. The stable isomer(s) in solution and their protonation products are shown.
Scheme 2: The hydrazone-based molecular systems that were analysed in this paper, each having different rotor...
In certain cases it has been shown that intramolecularly H-bonded hydrazones exist predominantly as the kinetically stable Z isomer in solution [27-29]. We were expecting that the intramolecular H-bonds in PPH-2 and PPH-3 would drive them to adopt the Z configuration in solution as well, leading to a low-field-lying NH signal (12–16 ppm) [22-24]. However, this is not the case with PPH-2. The hydrazone N–H proton in PPH-2 resonates at 8.95 ppm, which clearly shows that it is not H-bonded to the pyridyl nitrogen, indicating that the E configuration is the predominant isomer in solution (CD3CN). The addition of trifluoroacetic acid (TFA) only results in a general downfield shift of the aromatic and the hydrazone N–H proton signals as a result of protonation, which reaches saturation with 3 equiv of TFA. Unlike in PPH-1, signals from other species are not observed during the course of protonation, suggesting that the protonation of PPH-2 with TFA is a fast equilibrium, and that, as expected, it does not cause any isomerization. Similar to PPH-2, the 1H NMR spectrum of PPH-3 shows a signal for the hydrazone N–H proton at 8.24 ppm indicating that it too is in the E configuration. The protonation of PPH-3 with TFA is a fast equilibrium as well, without any indication of rotary motion (i.e., isomerization).
On the other hand, PPH-4, in which R is a strong electron-withdrawing group (–CN) shows two sets of signals in the 1H NMR spectrum (CD3CN), indicating that two isomers, having a 4:1 ratio, coexist in solution. The major isomer shows a hydrazone N–H signal at 9.60 ppm, indicating that it is the Z isomer, in which an intramolecular H-bond is not present. On the other hand, the hydrazone N–H signal of the minor isomer resonates at 15.12 ppm, which is characteristic of H-bonded N–H signals, suggesting that the minor isomer is actually the E configuration. Such an unusual E/Z isomer ratio was reported before for similar systems, and it was attributed to kinetic stability of the Z isomer, in addition to solvent effects [27-29]. The titration of PPH-4 with TFA only affects the major isomer (Z), while the minor isomer (E) remains intact even in the presence of 10 equiv of TFA. The changes in the 1H NMR spectrum of PPH-4-Z are similar to those of PPH-2 and PPH-3, except for the fact that it requires an excess of TFA (ca. 10 equiv) for the protonation to reach saturation. This observation can be attributed to the strong electron-withdrawing nature of the CN group, which drastically decreases the basicity of the pyridyl nitrogen. Furthermore, since the pyridyl nitrogen in PPH-4-E is H-bonded to the hydrazone N–H, the basicity of PPH-4-E becomes even lower, which explains why PPH-4-E does not become protonated even in the presence of 10 equiv of TFA.
Structurally, PPH-5 is the closest to PPH-1, that is, instead of an acyl ester group, PPH-5 has an acetyl residue as the R group. The 1H NMR spectrum of PPH-5 in CD3CN shows only one set of signals, and a sharp singlet at 14.54 ppm for the hydrazone N–H proton, indicating that it is H-bonded to the pyridyl nitrogen. Since the acetyl group is a less effective H-bond acceptor than ethyl ester, it is reasonable that PPH-5 exists exclusively in the E form in solution. When TFA is added to the solution, a second set of signals arises, which grows as the amount of acid increases. The protonation of the pyridyl ring results in the downfield shift of the aromatic signals, except for proton H1, which shifts from 8.92 to 8.70 ppm as it is no longer affected by the H-bond [22-24]. Moreover, the hydrazone N–H signal shifts to a higher field (13.22 ppm) in congruence with what is observed in PPH-1 [24]. These changes are consistent with those observed during the acid-activated switching of PPH-1, suggesting that PPH-5 switches from the E to the Z configuration upon protonation. However, the switching process of PPH-5 is relatively inefficient as there is still ca. 44% of PPH-5-E remaining in solution even when 30 equiv of TFA is added.
In order to rationalize the different behaviour of the structurally similar switches, PPH-1 and PPH-5, a quantitative evaluation of the thermodynamic process is necessary. Taking a look at the acid-activated switching process of PPH-1 (Scheme 1), we can formulate the following equations for the acid-induced E/Z isomerization:
![[1860-5397-8-98-i3]](/bjoc/content/inline/1860-5397-8-98-i3.svg?max-width=590&scale=1.18182)
![[1860-5397-8-98-i4]](/bjoc/content/inline/1860-5397-8-98-i4.svg?max-width=590&scale=1.18182)
![[1860-5397-8-98-i5]](/bjoc/content/inline/1860-5397-8-98-i5.svg?max-width=590&scale=1.18182)
where KP is the equilibrium constant of the protonation step, KI is the equilibrium constant for the rotation process, and KS is the overall equilibrium constant for the switching reaction. The pKa of PPH-1 is actually log10KP, so KS also equals ![[Graphic 1]](/bjoc/content/inline/1860-5397-8-98-i6.svg?max-width=637&scale=1.18182) . From the above equations, it becomes clear that KS can be used as an index to evaluate the feasibility of the switching process in hydrazone-based switches; the larger the KS value, the easier the switching process. In the case of PPH-1 versus PPH-5, since the acetyl group is a stronger electron-withdrawing group than the ester group, the basicity (pKa) of the pyridyl group in PPH-1 will be higher than in PPH-5. Moreover, the ester group is a better H-bond acceptor than the acetyl group, which means that the protonated Z configuration of PPH-1 is more stable than that of PPH-5, resulting in a larger KI for PPH-1. Thus, it can be qualitatively deduced that PPH-1 has a larger KS than PPH-5, suggesting that PPH-1 is a more ideal system to be used as a molecular switch. This analysis is clearly in line with the acid switching experiments that show that PPH-1 can be fully switched, whereas PPH-5 cannot.
. From the above equations, it becomes clear that KS can be used as an index to evaluate the feasibility of the switching process in hydrazone-based switches; the larger the KS value, the easier the switching process. In the case of PPH-1 versus PPH-5, since the acetyl group is a stronger electron-withdrawing group than the ester group, the basicity (pKa) of the pyridyl group in PPH-1 will be higher than in PPH-5. Moreover, the ester group is a better H-bond acceptor than the acetyl group, which means that the protonated Z configuration of PPH-1 is more stable than that of PPH-5, resulting in a larger KI for PPH-1. Thus, it can be qualitatively deduced that PPH-1 has a larger KS than PPH-5, suggesting that PPH-1 is a more ideal system to be used as a molecular switch. This analysis is clearly in line with the acid switching experiments that show that PPH-1 can be fully switched, whereas PPH-5 cannot.
Conclusion
In summary, we have synthesized four hydrazone-based systems having different R groups as part of the rotor section. The role of the R group was assessed vis-à-vis the switching of the system, and it was found that for the switch to operate effectively it is crucial that (1) the R group be able to offer a second H-bond-accepting site in order to provide enough driving force for the rotor to move; and (2) the R group be a moderate H-bond acceptor, otherwise the isomer generated will not be stable enough to enable full conversion (isomerization).
Supporting Information
| Supporting Information File 1: Experimental section and acid titration of the hydrazone compounds. | ||
| Format: PDF | Size: 371.4 KB | Download |
References
-
Schilwa, M., Ed. Molecular Motors; Wiley-VCH: Weinheim, Germany, 2003.
Return to citation in text: [1] -
Kelly, T. R.; De Silva, H.; Silva, R. A. Nature 1999, 401, 150–152. doi:10.1038/43639
Return to citation in text: [1] -
Leigh, D. A.; Wong, J. K. Y.; Dehez, F.; Zerbetto, F. Nature 2003, 424, 174–179. doi:10.1038/nature01758
Return to citation in text: [1] -
Fletcher, S. P.; Dumur, F.; Pollard, M. M.; Feringa, B. L. Science 2005, 310, 80–82. doi:10.1126/science.1117090
Return to citation in text: [1] -
Kelly, T. R.; Cai, X.; Damkaci, F.; Panicker, S. B.; Tu, B.; Bushell, S. M.; Cornella, I.; Piggott, M. J.; Salives, R.; Cavero, M.; Zhao, Y.; Jasmin, S. J. Am. Chem. Soc. 2007, 129, 376–386. doi:10.1021/ja066044a
Return to citation in text: [1] -
Feringa, B. L. J. Org. Chem. 2007, 72, 6635–6652. doi:10.1021/jo070394d
Return to citation in text: [1] -
Badjić, J. D.; Balzani, V.; Credi, A.; Silvi, S.; Stoddart, J. F. Science 2004, 303, 1845–1849. doi:10.1126/science.1094791
Return to citation in text: [1] -
Berná, J.; Leigh, D. A.; Lubomska, M.; Mendoza, S. M.; Pérez, E. M.; Rudolf, P.; Teobaldi, G.; Zerbetto, F. Nat. Mater. 2005, 4, 704–710. doi:10.1038/nmat1455
Return to citation in text: [1] -
Bonnet, S.; Collin, J.-P.; Koizumi, M.; Mobian, P.; Sauvage, J.-P. Adv. Mater. 2006, 18, 1239–1250. doi:10.1002/adma.200502394
Return to citation in text: [1] -
Wang, W.; Kaifer, A. E. Angew. Chem., Int. Ed. 2006, 45, 7042–7046. doi:10.1002/anie.200602220
Return to citation in text: [1] -
Nijhuis, C. A.; Ravoo, B. J.; Huskens, J.; Reinhoudt, D. N. Coord. Chem. Rev. 2007, 251, 1761–1780. doi:10.1016/j.ccr.2007.02.001
Return to citation in text: [1] -
Juluri, B. K.; Kumar, A. S.; Liu, Y.; Ye, T.; Yang, Y.-W.; Flood, A. H.; Fang, L.; Stoddart, J. F.; Weiss, P. S.; Huang, T. J. ACS Nano 2009, 3, 291–300. doi:10.1021/nn8002373
Return to citation in text: [1] -
Parimal, K.; Witlicki, E. H.; Flood, A. H. Angew. Chem., Int. Ed. 2010, 49, 4628–4632. doi:10.1002/anie.201001003
Return to citation in text: [1] -
Balzani, V.; Credi, A.; Venturi, M. Pure Appl. Chem. 2003, 75, 541–547. doi:10.1351/pac200375050541
Return to citation in text: [1] -
Kottas, G. S.; Clarke, L. I.; Horinek, D.; Michl, J. Chem. Rev. 2005, 105, 1281–1376. doi:10.1021/cr0300993
Return to citation in text: [1] -
Eelkema, R.; Pollard, M. M.; Vicario, J.; Katsonis, N.; Ramon, B. S.; Bastiaansen, C. W. M.; Broer, D. J.; Feringa, B. L. Nature 2006, 440, 163. doi:10.1038/440163a
Return to citation in text: [1] -
Muraoka, T.; Kinbara, K.; Aida, T. Nature 2006, 440, 512–515. doi:10.1038/nature04635
Return to citation in text: [1] -
Russew, M.-M.; Hecht, S. Adv. Mater. 2010, 22, 3348–3360. doi:10.1002/adma.200904102
Return to citation in text: [1] -
Kulago, A. A.; Mes, E. M.; Klok, M.; Meetsma, A.; Brouwer, A. M.; Feringa, B. L. J. Org. Chem. 2010, 75, 666–679. doi:10.1021/jo902207x
Return to citation in text: [1] -
Landaluce, T. F.; London, G.; Pollard, M. M.; Rudolf, P.; Feringa, B. L. J. Org. Chem. 2010, 75, 5323–5325. doi:10.1021/jo1006976
Return to citation in text: [1] [2] -
Lubbe, A. S.; Ruangsupapichat, N.; Caroli, G.; Feringa, B. L. J. Org. Chem. 2011, 76, 8599–8610. doi:10.1021/jo201583z
Return to citation in text: [1] -
Landge, S. M.; Aprahamian, I. J. Am. Chem. Soc. 2009, 131, 18269–18271. doi:10.1021/ja909149z
Return to citation in text: [1] [2] [3] -
Su, X.; Aprahamian, I. Org. Lett. 2011, 13, 30–33. doi:10.1021/ol102422h
Return to citation in text: [1] [2] [3] -
Landge, S. M.; Tkatchouk, E.; Benítez, D.; Lanfranchi, D. A.; Elhabiri, M.; Goddard, W. A., III; Aprahamian, I. J. Am. Chem. Soc. 2011, 133, 9812–9823. doi:10.1021/ja200699v
Return to citation in text: [1] [2] [3] [4] -
Su, X.; Robbins, T. F.; Aprahamian, I. Angew. Chem., Int. Ed. 2011, 50, 1841–1844. doi:10.1002/anie.201006982
Return to citation in text: [1] -
Yang, Y.; Su, X.; Carroll, C. N.; Aprahamian, I. Chem. Sci. 2012, 3, 610–613. doi:10.1039/c1sc00658d
Return to citation in text: [1] [2] -
Karabatsos, G. J.; Shapiro, B. L.; Vane, F. M.; Fleming, J. S.; Ratka, J. S. J. Am. Chem. Soc. 1963, 85, 2784–2788. doi:10.1021/ja00901a025
Return to citation in text: [1] [2] -
Mitchell, A. D.; Nonhebel, D. C. Tetrahedron Lett. 1975, 16, 3859–3862. doi:10.1016/S0040-4039(00)91297-8
Return to citation in text: [1] [2] -
Butler, R. N.; Johnson, S. M. J. Chem. Soc., Perkin Trans. 1 1984, 2109–2116. doi:10.1039/P19840002109
Return to citation in text: [1] [2]
| 14. | Balzani, V.; Credi, A.; Venturi, M. Pure Appl. Chem. 2003, 75, 541–547. doi:10.1351/pac200375050541 |
| 15. | Kottas, G. S.; Clarke, L. I.; Horinek, D.; Michl, J. Chem. Rev. 2005, 105, 1281–1376. doi:10.1021/cr0300993 |
| 16. | Eelkema, R.; Pollard, M. M.; Vicario, J.; Katsonis, N.; Ramon, B. S.; Bastiaansen, C. W. M.; Broer, D. J.; Feringa, B. L. Nature 2006, 440, 163. doi:10.1038/440163a |
| 17. | Muraoka, T.; Kinbara, K.; Aida, T. Nature 2006, 440, 512–515. doi:10.1038/nature04635 |
| 18. | Russew, M.-M.; Hecht, S. Adv. Mater. 2010, 22, 3348–3360. doi:10.1002/adma.200904102 |
| 22. | Landge, S. M.; Aprahamian, I. J. Am. Chem. Soc. 2009, 131, 18269–18271. doi:10.1021/ja909149z |
| 23. | Su, X.; Aprahamian, I. Org. Lett. 2011, 13, 30–33. doi:10.1021/ol102422h |
| 24. | Landge, S. M.; Tkatchouk, E.; Benítez, D.; Lanfranchi, D. A.; Elhabiri, M.; Goddard, W. A., III; Aprahamian, I. J. Am. Chem. Soc. 2011, 133, 9812–9823. doi:10.1021/ja200699v |
| 10. | Wang, W.; Kaifer, A. E. Angew. Chem., Int. Ed. 2006, 45, 7042–7046. doi:10.1002/anie.200602220 |
| 11. | Nijhuis, C. A.; Ravoo, B. J.; Huskens, J.; Reinhoudt, D. N. Coord. Chem. Rev. 2007, 251, 1761–1780. doi:10.1016/j.ccr.2007.02.001 |
| 12. | Juluri, B. K.; Kumar, A. S.; Liu, Y.; Ye, T.; Yang, Y.-W.; Flood, A. H.; Fang, L.; Stoddart, J. F.; Weiss, P. S.; Huang, T. J. ACS Nano 2009, 3, 291–300. doi:10.1021/nn8002373 |
| 13. | Parimal, K.; Witlicki, E. H.; Flood, A. H. Angew. Chem., Int. Ed. 2010, 49, 4628–4632. doi:10.1002/anie.201001003 |
| 24. | Landge, S. M.; Tkatchouk, E.; Benítez, D.; Lanfranchi, D. A.; Elhabiri, M.; Goddard, W. A., III; Aprahamian, I. J. Am. Chem. Soc. 2011, 133, 9812–9823. doi:10.1021/ja200699v |
| 7. | Badjić, J. D.; Balzani, V.; Credi, A.; Silvi, S.; Stoddart, J. F. Science 2004, 303, 1845–1849. doi:10.1126/science.1094791 |
| 8. | Berná, J.; Leigh, D. A.; Lubomska, M.; Mendoza, S. M.; Pérez, E. M.; Rudolf, P.; Teobaldi, G.; Zerbetto, F. Nat. Mater. 2005, 4, 704–710. doi:10.1038/nmat1455 |
| 9. | Bonnet, S.; Collin, J.-P.; Koizumi, M.; Mobian, P.; Sauvage, J.-P. Adv. Mater. 2006, 18, 1239–1250. doi:10.1002/adma.200502394 |
| 22. | Landge, S. M.; Aprahamian, I. J. Am. Chem. Soc. 2009, 131, 18269–18271. doi:10.1021/ja909149z |
| 23. | Su, X.; Aprahamian, I. Org. Lett. 2011, 13, 30–33. doi:10.1021/ol102422h |
| 24. | Landge, S. M.; Tkatchouk, E.; Benítez, D.; Lanfranchi, D. A.; Elhabiri, M.; Goddard, W. A., III; Aprahamian, I. J. Am. Chem. Soc. 2011, 133, 9812–9823. doi:10.1021/ja200699v |
| 2. | Kelly, T. R.; De Silva, H.; Silva, R. A. Nature 1999, 401, 150–152. doi:10.1038/43639 |
| 3. | Leigh, D. A.; Wong, J. K. Y.; Dehez, F.; Zerbetto, F. Nature 2003, 424, 174–179. doi:10.1038/nature01758 |
| 4. | Fletcher, S. P.; Dumur, F.; Pollard, M. M.; Feringa, B. L. Science 2005, 310, 80–82. doi:10.1126/science.1117090 |
| 5. | Kelly, T. R.; Cai, X.; Damkaci, F.; Panicker, S. B.; Tu, B.; Bushell, S. M.; Cornella, I.; Piggott, M. J.; Salives, R.; Cavero, M.; Zhao, Y.; Jasmin, S. J. Am. Chem. Soc. 2007, 129, 376–386. doi:10.1021/ja066044a |
| 6. | Feringa, B. L. J. Org. Chem. 2007, 72, 6635–6652. doi:10.1021/jo070394d |
| 27. | Karabatsos, G. J.; Shapiro, B. L.; Vane, F. M.; Fleming, J. S.; Ratka, J. S. J. Am. Chem. Soc. 1963, 85, 2784–2788. doi:10.1021/ja00901a025 |
| 28. | Mitchell, A. D.; Nonhebel, D. C. Tetrahedron Lett. 1975, 16, 3859–3862. doi:10.1016/S0040-4039(00)91297-8 |
| 29. | Butler, R. N.; Johnson, S. M. J. Chem. Soc., Perkin Trans. 1 1984, 2109–2116. doi:10.1039/P19840002109 |
| 25. | Su, X.; Robbins, T. F.; Aprahamian, I. Angew. Chem., Int. Ed. 2011, 50, 1841–1844. doi:10.1002/anie.201006982 |
| 26. | Yang, Y.; Su, X.; Carroll, C. N.; Aprahamian, I. Chem. Sci. 2012, 3, 610–613. doi:10.1039/c1sc00658d |
| 22. | Landge, S. M.; Aprahamian, I. J. Am. Chem. Soc. 2009, 131, 18269–18271. doi:10.1021/ja909149z |
| 23. | Su, X.; Aprahamian, I. Org. Lett. 2011, 13, 30–33. doi:10.1021/ol102422h |
| 24. | Landge, S. M.; Tkatchouk, E.; Benítez, D.; Lanfranchi, D. A.; Elhabiri, M.; Goddard, W. A., III; Aprahamian, I. J. Am. Chem. Soc. 2011, 133, 9812–9823. doi:10.1021/ja200699v |
| 27. | Karabatsos, G. J.; Shapiro, B. L.; Vane, F. M.; Fleming, J. S.; Ratka, J. S. J. Am. Chem. Soc. 1963, 85, 2784–2788. doi:10.1021/ja00901a025 |
| 28. | Mitchell, A. D.; Nonhebel, D. C. Tetrahedron Lett. 1975, 16, 3859–3862. doi:10.1016/S0040-4039(00)91297-8 |
| 29. | Butler, R. N.; Johnson, S. M. J. Chem. Soc., Perkin Trans. 1 1984, 2109–2116. doi:10.1039/P19840002109 |
| 20. | Landaluce, T. F.; London, G.; Pollard, M. M.; Rudolf, P.; Feringa, B. L. J. Org. Chem. 2010, 75, 5323–5325. doi:10.1021/jo1006976 |
| 19. | Kulago, A. A.; Mes, E. M.; Klok, M.; Meetsma, A.; Brouwer, A. M.; Feringa, B. L. J. Org. Chem. 2010, 75, 666–679. doi:10.1021/jo902207x |
| 20. | Landaluce, T. F.; London, G.; Pollard, M. M.; Rudolf, P.; Feringa, B. L. J. Org. Chem. 2010, 75, 5323–5325. doi:10.1021/jo1006976 |
| 21. | Lubbe, A. S.; Ruangsupapichat, N.; Caroli, G.; Feringa, B. L. J. Org. Chem. 2011, 76, 8599–8610. doi:10.1021/jo201583z |
| 26. | Yang, Y.; Su, X.; Carroll, C. N.; Aprahamian, I. Chem. Sci. 2012, 3, 610–613. doi:10.1039/c1sc00658d |
© 2012 Su et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)