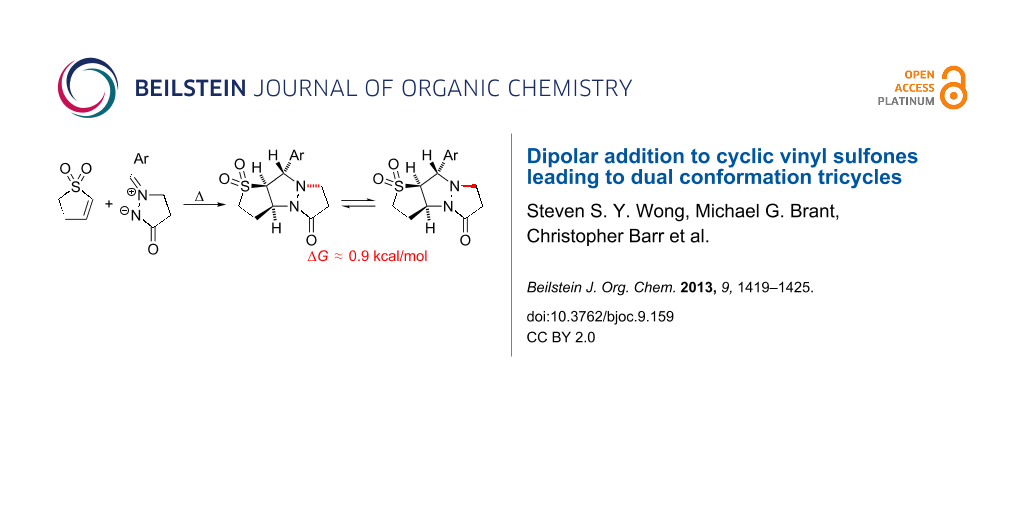
Abstract
Dipolar addition of cyclic azomethine imines with cyclic vinyl sulfones gave rise to functionalized tricycles that exhibited fluxional behavior in solution at room temperature. The scope of the synthetic methodology was explored, and the origin of the fluxional behavior was probed by NMR methods together with DFT calculations. This behavior was ultimately attributed to stereochemical inversion at one of two nitrogen centers embedded in the tricyclic framework. Two tetracycles were also synthesized, and the degree of signal-broadening in the NMR spectra was found to depend on the presence of substitution next to the inverting nitrogen center.

Graphical Abstract
Introduction
The 1,3-dipolar cycloaddition [1-3] represents a powerful methodology for the expedient regio- and stereospecific synthesis of five-membered ring N-, O- or S-containing heterocycles [4,5]. Various 1,3-dipoles can be used, including nitrones [6], azomethine ylides [7], diazoalkanes [8,9] and many others [10-12]. α,β-Unsaturated carbonyl compounds are often found to be good coupling partners for these cycloadditions, and in many cases the reaction rate can be enhanced through the addition of a Lewis acid that can coordinate to the carbonyl function, thereby lowering the energy of the olefin LUMO. By contrast, vinyl sulfones (with the exception of simple aryl vinyl sulfone species [13-21]) have been less-frequently employed as acceptors in 1,3-dipolar cycloadditions [22-25]. Indeed, we are only aware of a single prior example of a cyclic alkyl vinyl sulfone participating in a dipolar addition with an azomethine ylide [26]. In part, this lack of reactivity is due to a poor orbital overlap between the sulfone and olefin functions [27-29]; while vinyl sulfones are electrophilic (due to the inductive effects of the sulfone group), their LUMO cannot be as easily perturbed by the addition of a Lewis acid.
We previously described the synthesis of a family of [3.3.0] bicyclic vinyl sulfones starting from 3-sulfolene [30], and their elaboration to inhibitors of viral neuraminidase (Scheme 1) [31]. Understanding the conformational preferences of the underlying bicyclic structure was crucial to the design and assembly of such inhibitors. In keeping with our interest in the synthesis and utilization of conformationally constrained polycyclic sulfones, we sought to explore the cycloaddition between 2-sulfolene (1a) [32] and 1,3-dipoles (2, Table 1, see below) derived from 3-pyrazolidinone [33]. Reaction of 2 with acetylenic sulfones has been reported [34], but cyclic alkyl vinyl sulfones have not previously been demonstrated as coupling partners for 2.
![[1860-5397-9-159-i1]](/bjoc/content/inline/1860-5397-9-159-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of a conformationally constrained bicyclic sulfone, and application as an inhibitor of an enzyme target.
Scheme 1: Synthesis of a conformationally constrained bicyclic sulfone, and application as an inhibitor of an...
Results and Discussion
A brief survey of reaction conditions revealed that dipole 2b (easily prepared by condensing 3-pyrazolidinone with benzaldehyde) [33] could be reacted with 1a in DMSO under reflux. When the two reagents were combined in equimolar quantities (Table 1, entry 2), the desired coupling product 3b was isolated in 38% yield. Dipole 2 is known to be capable of homo-dimerization [35], and examination of the crude NMR spectra for the reaction revealed the presence of this dimer along with unreacted 1a. We were gratified to find that simply increasing the amount of dipole to 3 equiv amplified the yield to 86%.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-159-i2.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-159-i3.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-159-i4.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-159-i5.svg?max-width=637&scale=1.0)
Seeking to better understand how electronic properties in the dipole affected the reaction outcome, we repeated these coupling reactions using both electron rich (2a, Table 1, entry 1) and electron poor (2c, Table 1, entry 3) dipoles. When 1 equiv of dipole was used, a clear trend emerged wherein greater electron density was associated with an increased yield of product. At first glance this might seem counterintuitive, since more electron density at the hydrazone function should disfavor formation of one of the new C–C bonds in 3. However, we speculate that the increase in electron density serves to disfavor dimerization more than 1,3-dipolar addition to 1a. As a result, more dipole remains in solution to undergo the desired coupling. The effect of this change was somewhat muted when 3 equiv of dipole were used, resulting in synthetically useful yields of all three tricycles.
Characterization of the products 3a–3c was complicated by the presence of very broad signals in both the 1H and 13C NMR spectra. Interestingly, however, HSQC analysis revealed that these broadened signals did not correlate with one another (i.e., one of the very broad 1H signals correlated to a sharp 13C signal, while one of the broad 13C signals correlated to a sharp signal in the 1H NMR spectrum). These observations suggested that signal broadening was not due to quadrupolar effects from the inclusion of the two nitrogen atoms (in which case broadening would be expected to be localized to certain regions of the molecular structure) but was more likely to be due to conformational switching of the molecule on a timescale similar to that of the NMR experiment.
We therefore turned to variable temperature NMR spectroscopy, both to assist in confirming the structures of the products, and to elucidate the conformational changes that were occurring. Extensive 1D and 2D NMR analysis of 3a and 3b at 353 K in DMSO-d6 (where sharp signals were observed for both compounds) confirmed the regio- and stereochemical outcome of the cycloaddition as that indicated for structure 3. This structural analysis was subsequently confirmed by X-ray crystallographic analysis of 3a (Figure 1) [36].
![[1860-5397-9-159-1]](/bjoc/content/figures/1860-5397-9-159-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Further VT NMR studies of 3a in CDCl3 (Figure 2B) revealed a substantial broadening of several key 1H NMR signals below 330 K, with a coalescence temperature of approximately 280 K. At lower temperatures, two distinct conformations could be identified; by 213 K these were sufficiently well-resolved to permit integration (revealing a ≈7:1 ratio of the two conformers) and more extensive NMR analysis (HSQC, HMBC, ROESY, TOCSY and EXSY).
![[1860-5397-9-159-2]](/bjoc/content/figures/1860-5397-9-159-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Assignment of major and minor conformations of 3a; A: DFT-calculated conformers; B: Collected 1H NMR spectra at various temperatures, in CDCl3. Assignments in Figure 2B refer to the structures in Figure 2A, where blue = major isomer, red = minor isomer and purple = equilibrating mixture.
Figure 2: Assignment of major and minor conformations of 3a; A: DFT-calculated conformers; B: Collected 1H NM...
The most significant difference between the NMR data for the two conformations was the substantial upfield shift for the two protons adjacent to the carbonyl (‘d’ in Figure 2) in the major conformer. This was consistent with this methylene group being substantially shielded by the electron-rich aromatic ring, as would be expected for the conformation observed in the X-ray structure (Figure 1). This upfield shift was not present in the minor conformer, suggesting that the pyrazolidinone ring had moved away from the aromatic ring’s magnetic influence.
Analysis of several lines of evidence (including nOe information from the NMR studies, construction of likely molecular models, and comparison with known fluxional behavior for related structures [37]) strongly suggested that these differences could best be explained by the tricycle “breathing” through stereochemical inversion at the nitrogen atom distal to the carbonyl. To test this hypothesis, we carried out DFT calculations [38] on structures in which the pyrazolidinone and sulfolane rings were oriented syn or anti to one another (i.e., “endo” or “exo” conformations, respectively). Geometry optimization resulted in the two lowest-energy structures shown in Figure 2A. Compellingly, the difference in ΔHf between these two structures (0.93 kcal/mol in favor of the exo conformer) agrees very well with the experimental difference in energy calculated from NMR integration of the two conformations at 213 K (0.8 kcal/mol).
The calculated structure of the lower-energy exo conformer was similar to the solid-state structure shown in Figure 1, and was consistent with the nOe and NMR shielding data discussed above. Seeking further evidence linking the calculated exo and endo structures to the observed major and minor conformers, we calculated the 13C NMR shifts for our geometry-optimized structures, and compared these to the shifts obtained experimentally for the two conformations (Table 2). These agreed exceptionally well for those carbons not connected to the sulfone (relativistic effects were not well handled by our computational method; shifts for carbons close to the sulfone could therefore not be accurately calculated).
Of particular note is the effect of the nitrogen inversion on carbons “d” and “e”. In the exo conformer, these atoms are brought into proximity with the shielding aromatic ring, resulting in atypically upfield chemical shifts. This shielding effect is not present in the endo conformer, and so these 13C shifts return to more typical values. The difference is substantial: 6 ppm for carbon “d” and 8 ppm for carbon “e”. The excellent agreement between theory and experiment for these two carbons in both conformations, together with good qualitative agreement between calculated and observed 1H NMR shifts (not shown) and other data summarized above, allows us to unambiguously assign the major conformer as exo, and the minor conformer as endo.
We next sought to evaluate whether the method could be extended toward the preparation of the analogous tetracycles, through the use of bicyclic vinyl sulfones as substrates. To this end, we attempted to react dipoles 2a and 2b with the readily prepared [39] bicycle 1b (Table 3, entry 1). Unfortunately, no cycloaddition adducts were observed even when additional heating was employed, either thermally in a sealed tube or in a microwave reactor.
Table 3: Cycloadditions with bicyclic sulfones, and related control experiments.a
| entry | sulfone 1 | dipole | product (yield) |
|---|---|---|---|
| 1 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-159-i6.svg?max-width=637&scale=1.0)
1b |
2a or 2b | – |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-159-i7.svg?max-width=637&scale=1.0)
1c |
2a |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-159-i8.svg?max-width=637&scale=1.0)
4 (37%) |
| 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-159-i9.svg?max-width=637&scale=1.0)
1d |
2a |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-159-i10.svg?max-width=637&scale=1.0)
5 (68%) |
| 4 | 1c |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-159-i11.svg?max-width=637&scale=1.0)
6 |
4 (67%) |
| 5b | 1c | 6 | 4 (70%) |
| 6c | 1c | 2b |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-159-i12.svg?max-width=637&scale=1.0)
7 (95%) |
a3 equiv of 2 or 6 were used for each experiment. b25 equiv of paraformaldehyde were added. cReaction was carried out in bromobenzene under reflux.
Bicycle 1b is somewhat more hindered than 1a, which is presumably the reason behind its lack of reactivity. In an attempt to attenuate the steric hindrance, we next examined the reaction of 2a with bicycle 1c (Table 3, entry 2). We were pleased to observe a tetracyclic product in this case (albeit in modest yield) but were surprised to find that the anisole ring was not included in the isolated compound. Indeed, adduct 4 appeared to result from transiminization of dipole 2a with formaldehyde (generated by the thermal decomposition of DMSO [40]), followed by selective cycloaddition of the more reactive dipole species. This suggests that 2a may be too hindered to react easily with bicycle 1c, but that this hindrance can be mitigated by in situ exchange to generate a dipole lacking the aromatic ring. Compound 4 displayed sharp signals in the 1H and 13C NMR spectra, presumably due to the lack of an aromatic substituent next to the sp3-hybridized nitrogen atom. Electronically, aryl vinyl sulfone 1c is unlike the alkyl sulfones 1a and 1b. To confirm that its aberrant reactivity was not due to electronic factors, we carried out the cycloadditon of 2a with the simpler acyclic aryl vinyl sulfone 1d (Table 3, entry 3). As anticipated, this reaction proceeded smoothly to afford the expected product 5 in reasonable yield. To confirm that the anomalous methylene group in product 4 originated from the decomposition of DMSO, we reacted bicycle 1c with 3-pyrazolidinone (6). Once again we isolated tetracycle 4, this time in a considerably improved yield of 67% (Table 3, entry 4). Adding an excess of paraformaldehyde to the reaction mixture (Table 3, entry 5) had no significant effect on the yield. Finally, to explore the dipolar addition in the absence of competing transiminization processes, we reacted bicycle 1c with 3 equivalents of 2b, in refluxing bromobenzene (Table 3, entry 6). This provided an excellent yield of the desired tetracycle 7, confirming that even hindered dipolar additions of this type can proceed if no alternative pathways are open to the reactants. The additional aromatic substituent in 7 (relative to 4) once again contributed to significant broadening in both the 1H and 13C NMR spectra.
Conclusion
Our results comprise the first syntheses of tricyclic sulfones by 1,3-dipolar cycloadditions of azomethine imines with cyclic alkyl vinyl sulfones. Only single diastereomers were obtained in each case, and the yields were respectable when an excess of the dipole was used. Both electron-donating and electron-withdrawing substituents on the dipole’s aromatic function were tolerated. Initial attempts to synthesize a tetracyclic analogue resulted in the appearance of compound 4, resulting from an unexpected transimination/cycloaddition sequence. By avoiding the transimination pathway, the original target tetracycle 7 could be isolated.
Of potentially greater interest than the syntheses themselves, tricycles 3a–3c (as well as tetracycle 7) exhibited an unusual fluxional behavior at room temperature, which was ultimately traced to stereochemical inversion at the sp3-hybridized nitrogen. This behavior is consistent with previous examples of fluxional alkaloids, but represents a new structural type that may find application in supramolecular or medicinal chemistry. DFT- and NMR-based studies agreed remarkably well in implicating the two interconverting species as the “endo” and “exo” conformers shown in Figure 2.
Experimental
General methods. Unless otherwise stated, all reagents were purchased from commercial suppliers and used without further purification. DMSO was dried by distillation over CaH2. 1H chemical shifts are reported in parts per million (ppm, δ scale) downfield from tetramethylsilane, and are referenced to residual protium in the NMR solvent (CDCl3: δ 7.26). Likewise, 13C chemical shifts are referenced to the carbon resonances of the solvent (CDCl3: δ 77.00). TLC plates were visualized by exposure to KMnO4 stain. Accurate masses were obtained using an orbitrap MS. Infrared spectra were collected using an FT IR spectrometer.
General procedure for the dipolar cycloaddition. The vinyl sulfone (0.5 mmol) and dipole (3 equiv) were combined in dry DMSO (2.0 mL), and the resulting solution was heated under reflux in a sand bath. After 24 h, the solution was concentrated under vacuum at 50 °C. The remaining black viscous oil was purified by flash-column chromatography using a 2–10% gradient of methanol in dichloromethane, to afford the desired adduct as a brown solid.
Tricycle 3a: mp 189–192 °C; IR (film, cm−1): 1694, 1514, 1302, 1254, 1112; 1H NMR (CDCl3, 300 MHz, rt) δ 7.17–7.07 (m, 2H), 6.92 (d, J = 8.8 Hz, 2H), 5.01 (br s, 1H), 4.46 (br s, 1H), 3.81 (s, 3H), 3.75 (dd, J = 7.8, 1.9 Hz, 1H), 3.34–3.08 (m, 4H), 2.80 (br s, 1H), 2.59–2.27 (m, 2H), 1.80 (br s, 1H); 13C NMR (CDCl3, 75 MHz, rt) δ 160.3 (C), 129.7 (CH), 125.9 (C), 114.7 (CH), 70.4 (br CH), 69.0 (br CH), 55.3 (CH), 55.2 (CH3), 48.2 (CH2), 44.6 (br, CH2), 32.8 (br, CH2), 25.8 (br, CH2); HRMS–ESI (m/z): calcd for [C15H18N2O4S]+, 322.0987; found, 322.0983.
Tricycle 3b: mp 134–137 °C; IR (film, cm−1): 1693, 1303, 1114; 1H NMR (CDCl3, 300 MHz, rt) δ 7.43–7.37 (m, 3H), 7.27–7.20 (m, 2H), 5.00 (br s, 1H), 4.46 (br s, 1H), 3.79 (dd, J = 7.8, 2.6 Hz, 1H), 3.33–3.12 (m, 4H), 2.87 (br s, 1H), 2.58–2.34 (m, 2H), 2.01 (br s, 1H); 13C NMR (CDCl3, 75 MHz, rt) δ 134.3 (C), 129.2 (CH), 129.2 (CH), 128.3 (CH), 70.5 (CH), 69.3 (CH), 55.2 (CH), 48.1 (CH2), 44.9 (br, CH2), 33.4 (br, CH2), 25.4 (br, CH2); HRMS–ESI (m/z): calcd for [C14H16N2O3S]+, 292.0882; found, 292.0883.
Tricycle 3c: mp 201–203 °C; IR (film, cm−1): 1694, 1520, 1349, 1305, 1113; 1H NMR (CDCl3, 300 MHz, rt) δ 8.27 (d, J = 8.8 Hz, 2H), 7.61 (d, J = 8.6 Hz, 2H), 4.81 (br t, J = 6.4 Hz, 1H), 4.32 (br d, J = 4.9 Hz, 1H), 3.69 (dd, J = 7.8, 5.8 Hz, 1H), 3.40–2.94 (m, 5H), 2.77–2.36 (m, 3H); 13C NMR (CDCl3, 75 MHz, rt) δ 148.4 (C), 142.4 (C), 128.9 (CH), 124.4 (CH), 69.6 (CH), 55.3 (CH), 47.9 (CH2); HRMS–ESI (m/z): calcd for [C14H15N3O5S]+, 337.0732; found, 337.0730.
Tetracycle 4: oil; IR (film, cm−1): 1713, 1305, 1149; 1H NMR (CDCl3, 300 MHz, rt) δ 7.79–7.56 (m, 4H), 5.89 (d, J = 8.1 Hz, 1H), 4.17 (t, J = 7.6 Hz, 1H), 3.94 (d, J = 10.4 Hz, 1H), 3.62 (dt, J = 12.2, 9.2 Hz, 1H), 3.19 (ddd, J = 12.2, 9.2, 4.6 Hz, 1H), 2.87 (dd, J = 10.2, 7.7 Hz, 1H), 2.78 (dt, J = 17.8, 9.1 Hz, 1H) 2.66 (ddd, J = 17.8, 9.4, 4.6 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 175.7 (C), 139.4 (C), 134.7 (C), 134.4 (CH), 130.9 (CH), 128.0 (CH), 121.0 (CH), 64.1 (CH), 56.6 (CH), 55.3 (CH2), 46.1 (CH2), 30.1 (CH2); HRMS–ESI (m/z): calcd for [C12H12N2O3S + H]+, 265.0642; found 265.0640.
Bicycle 5: mp 184–186 °C; IR (film, cm−1): 1708, 1515, 1307, 1251, 1149; 1H NMR (CDCl3, 300 MHz, rt) δ 7.80, (d, J = 7.3 Hz, 2H), 7.60 (t, J = 7.4 Hz, 1H), 7.48 (t, J = 7.6 Hz, 2H), 7.25 (d, J = 8.7 Hz, 2H), 6.81 (d, J = 8.7 Hz, 2H), 4.35 (dd, J = 12.8, 3.7 Hz, 1H), 4.05–3.95 (m, 2H), 3.79 (s, 3H), 3.54 (dd, J = 12.6, 8.0 Hz, 1H), 3.47 (dt, J = 11.9, 9.1 Hz, 1H), 3.10 (ddd, J = 12.0, 9.5, 4.7 Hz, 1H), 2.80 (dt, J = 17.5, 8.9 Hz, 1H), 2.61 (ddd, J = 17.5, 9.3, 4.8 Hz, 1H); 13C NMR (CDCl3, 75 MHz, rt) δ 174.2 (C), 160.1 (C), 137.5 (C), 134.2 (CH), 129.4 (CH), 128.6 (CH), 127.3 (C), 114.3 (CH), 72.0 (CH), 68.5 (CH), 55.3 (CH3), 45.3 (CH2), 42.6 (CH2), 30.0 (CH2); HRMS–ESI (m/z): calcd for [C19H20N2O4S + H]+, 373.1217; found 373.1214.
Tetracycle 7: oil; IR (film, cm−1): 1698, 1304, 1150; 1H NMR (CDCl3, 300 MHz, rt) δ 7.96–7.84 (m, 1H), 7.81 (d, J = 7.7 Hz, 1H), 7.73 (td, J = 7.5, 1.2 Hz, 1H), 7.64 (br t, J = 7.7, 1H), 7.47–7.40 (m, 3H), 7.32–7.22 (m, 2H), 6.06 (d, J = 8.1 Hz, 1H), 4.78 (br s, 1H), 4.42 (dd, J = 8.2, 2.5 Hz, 1H), 3.34–3.17 (m, 1H), 3.15–3.02 (m, 1H), 2.36–2.18 (m, 1H), 1.94–1.62 (m, 1H); 13C NMR (CDCl3, 75 MHz) δ 172.4 (C), 138.9 (C), 134.5 (CH), 134.2 (C), 134.1 (C), 131.0 (CH), 129.4 (CH), 129.4 (CH), 128.9 (CH), 121.2 (CH), 70.1 (CH), 68.1 (CH), 56.1 (CH), 43.8 (CH2), 32.6 (CH2); HRMS–ESI (m/z): calcd for [C18H16N2O3S + Na]+, 363.0774; found 363.0771.
Supporting Information
Copies of 1H, 13C and DEPT-135 spectra for all new compounds, VT-NMR data and 2D NMR spectra for 3a, crystallographic data for 3a, and absolute energies and Cartesian coordinates for calculated conformers.
| Supporting Information File 1: Detailed measurement data. | ||
| Format: PDF | Size: 2.4 MB | Download |
Acknowledgements
We gratefully acknowledge the Canadian Institutes of Health Research for operating funds, as well as the University of Victoria Faculty of Science for a Summer Undergraduate Research Fellowship to S. W., and the Canada Research Chairs and Michael Smith Foundation for Health Research programs for salary support to J. W. We would also like to thank the Office of the Vice President of Research at the University of Notre Dame for financial support for the purchase of the copper microfocus source used in this research, as well as Ori Granot and the UVic Genome BC Proteomics Centre for support with the mass spectrometry.
References
-
Buchner, E. Ber. Dtsch. Chem. Ges. 1888, 21, 2637–2647. doi:10.1002/cber.18880210283
Return to citation in text: [1] -
Buchner, E.; Fritsch, M.; Papendieck, A.; Witter, H. Justus Liebigs Ann. Chem. 1893, 273, 214–231. doi:10.1002/jlac.18932730207
Return to citation in text: [1] -
Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565–598. doi:10.1002/anie.196305651
Return to citation in text: [1] -
Gothelf, K. V.; Jørgensen, K. A. Chem. Rev. 1998, 98, 863–910. doi:10.1021/cr970324e
Return to citation in text: [1] -
Padwa, A. 1,3-Dipolar Cycloadditions; John Wiley: New York, NY, 1984.
Return to citation in text: [1] -
Frederickson, M. Tetrahedron 1997, 53, 403–425. doi:10.1016/S0040-4020(96)01095-2
Return to citation in text: [1] -
Adrio, J.; Carretero, J. C. Chem. Commun. 2011, 47, 6784–6794. doi:10.1039/c1cc10779h
Return to citation in text: [1] -
Mish, M. R.; Guerra, F. M.; Carreira, E. M. J. Am. Chem. Soc. 1997, 119, 8379–8380. doi:10.1021/ja971708p
Return to citation in text: [1] -
Kanemasa, S.; Kanai, T. J. Am. Chem. Soc. 2000, 122, 10710–10711. doi:10.1021/ja002670a
Return to citation in text: [1] -
Huisgen, R. J. Org. Chem. 1976, 41, 403–419. doi:10.1021/jo00865a001
Return to citation in text: [1] -
Zhu, X.-F.; Henry, C. E.; Kwon, O. Tetrahedron 2005, 61, 6276–6282. doi:10.1016/j.tet.2005.03.104
Return to citation in text: [1] -
Partridge, K. M.; Guzei, I. A.; Yoon, T. P. Angew. Chem., Int. Ed. 2010, 49, 930–934. doi:10.1002/anie.200905801
Return to citation in text: [1] -
Clark, R. B.; Pearson, W. H. Org. Lett. 1999, 1, 349–352. doi:10.1021/ol990677v
Return to citation in text: [1] -
Huscroft, I. T.; Carlson, E. J.; Chicchi, G. G.; Kurtz, M. M.; London, C.; Raubo, P.; Wheeldon, A.; Kulagowski, J. J. Bioorg. Med. Chem. Lett. 2006, 16, 2008–2012. doi:10.1016/j.bmcl.2005.12.069
Return to citation in text: [1] -
Peese, K. M.; Gin, D. Y. Chem.–Eur. J. 2008, 14, 1654–1665. doi:10.1002/chem.200701290
Return to citation in text: [1] -
Kudryavtstev, K. V.; Ivantcova, P. M.; Churakov, A. V.; Vasin, V. A. Tetrahedron Lett. 2012, 53, 4300–4303. doi:10.1016/j.tetlet.2012.05.160
Return to citation in text: [1] -
Llamas, T.; Arrayás, R. G.; Carretero, J. C. Org. Lett. 2006, 8, 1795–1798. doi:10.1021/ol060314c
Return to citation in text: [1] -
Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Chem.–Eur. J. 2011, 17, 14224–14233. doi:10.1002/chem.201101606
Return to citation in text: [1] -
Tong, M.-C.; Li, J.; Tao, H.-Y.; Li, Y.-X.; Wang, C.-J. Chem.–Eur. J. 2011, 17, 12922–12927. doi:10.1002/chem.201102430
Return to citation in text: [1] -
Yamashita, Y.; Imaizumi, T.; Kobayashi, S. Angew. Chem., Int. Ed. 2011, 50, 4893–4896. doi:10.1002/anie.201008272
Return to citation in text: [1] -
González-Esguevillas, M.; Adrio, J.; Carretero, J. C. Chem. Commun. 2012, 48, 2149–2151. doi:10.1039/c2cc17149j
Return to citation in text: [1] -
Barr, D. A.; Donegan, G.; Grigg, R. J. Chem. Soc., Perkin Trans. 1 1989, 1550–1551. doi:10.1039/P19890001550
Return to citation in text: [1] -
Jones, R. C. F.; Rafiq, S.; Elsegood, M. R. J.; McKee, V.; Slater, M. J. Chem.–Asian J. 2010, 5, 461–465. doi:10.1002/asia.200900547
Return to citation in text: [1] -
Lakshmi, N. V.; Thirumurugan, P.; Jayakumar, C.; Perumal, P. T. Synlett 2010, 955–961. doi:10.1055/s-0029-1219550
Return to citation in text: [1] -
Atienza, R. L.; Roth, H. S.; Scheidt, K. A. Chem. Sci. 2011, 2, 1772–1776. doi:10.1039/c1sc00194a
Return to citation in text: [1] -
Yarmolchuk, V. S.; Mukan, I. L.; Grygorenko, O. O.; Tolmachev, A. A.; Shishkina, S. V.; Shishkin, O. V.; Komarov, I. V. J. Org. Chem. 2011, 76, 7010–7016. doi:10.1021/jo200878t
Return to citation in text: [1] -
Müller, C.; Schweig, A. Tetrahedron 1973, 29, 3973–3977. doi:10.1016/0040-4020(73)80222-4
Return to citation in text: [1] -
Caramella, P.; Albini, E.; Bandiera, T.; Coda, A. C.; Grünanger, P.; Albini, F. M. Tetrahedron 1983, 39, 689–699. doi:10.1016/S0040-4020(01)91846-0
Return to citation in text: [1] -
Barzaghi, M.; Beltrame, P. L.; Croce, P. D.; Buttero, P. D.; Licandro, E.; Maiorana, S.; Zecchi, G. J. Org. Chem. 1983, 48, 3807–3810. doi:10.1021/jo00169a041
Return to citation in text: [1] -
Brant, M. G.; Bromba, C. M.; Wulff, J. E. J. Org. Chem. 2010, 75, 6312–6315. doi:10.1021/jo101389c
Return to citation in text: [1] -
Brant, M. G.; Wulff, J. E. Org. Lett. 2012, 14, 5876–5879. doi:10.1021/ol3027939
Return to citation in text: [1] -
Bailey, W. J.; Cummins, E. W. J. Am. Chem. Soc. 1954, 76, 1932–1936. doi:10.1021/ja01636a058
Return to citation in text: [1] -
Xin, Y.; Zhao, J.; Gu, J.; Zhu, S. J. Fluorine Chem. 2011, 132, 402–408. doi:10.1016/j.jfluchem.2011.03.023
Return to citation in text: [1] [2] -
Gao, D.; Zhai, H.; Parvez, M.; Back, T. G. J. Org. Chem. 2008, 73, 8057–8068. doi:10.1021/jo801621d
Return to citation in text: [1] -
Dorn, H.; Kreher, T. Tetrahedron Lett. 1988, 29, 2939–2942. doi:10.1016/0040-4039(88)85052-4
Return to citation in text: [1] -
CCDC 939241 contains the supplementary crystallographic data for compound 3a. This data is available from the Cambridge Crystallographic Data Centre.
Return to citation in text: [1] -
Bosque, I.; González-Gómez, J. C.; Guijarro, A.; Foubelo, F.; Yus, M. J. Org. Chem. 2012, 77, 10340–10346. doi:10.1021/jo302045y
Return to citation in text: [1] -
All calculations were performed at the B3LYP/6-31G* level and were done using the Spartan 06 Molecular Modeling package.
Return to citation in text: [1] -
Chou, T.-s.; Chang, L.-J.; Tso, H.-H. J. Chem. Soc., Perkin Trans. 1 1986, 1039–1042. doi:10.1039/P19860001039
Return to citation in text: [1] -
Traynelis, V. J.; Hergenrother, W. L. J. Org. Chem. 1964, 29, 221–222. doi:10.1021/jo01024a505
Return to citation in text: [1]
| 37. | Bosque, I.; González-Gómez, J. C.; Guijarro, A.; Foubelo, F.; Yus, M. J. Org. Chem. 2012, 77, 10340–10346. doi:10.1021/jo302045y |
| 35. | Dorn, H.; Kreher, T. Tetrahedron Lett. 1988, 29, 2939–2942. doi:10.1016/0040-4039(88)85052-4 |
| 36. | CCDC 939241 contains the supplementary crystallographic data for compound 3a. This data is available from the Cambridge Crystallographic Data Centre. |
| 1. | Buchner, E. Ber. Dtsch. Chem. Ges. 1888, 21, 2637–2647. doi:10.1002/cber.18880210283 |
| 2. | Buchner, E.; Fritsch, M.; Papendieck, A.; Witter, H. Justus Liebigs Ann. Chem. 1893, 273, 214–231. doi:10.1002/jlac.18932730207 |
| 3. | Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565–598. doi:10.1002/anie.196305651 |
| 8. | Mish, M. R.; Guerra, F. M.; Carreira, E. M. J. Am. Chem. Soc. 1997, 119, 8379–8380. doi:10.1021/ja971708p |
| 9. | Kanemasa, S.; Kanai, T. J. Am. Chem. Soc. 2000, 122, 10710–10711. doi:10.1021/ja002670a |
| 34. | Gao, D.; Zhai, H.; Parvez, M.; Back, T. G. J. Org. Chem. 2008, 73, 8057–8068. doi:10.1021/jo801621d |
| 7. | Adrio, J.; Carretero, J. C. Chem. Commun. 2011, 47, 6784–6794. doi:10.1039/c1cc10779h |
| 33. | Xin, Y.; Zhao, J.; Gu, J.; Zhu, S. J. Fluorine Chem. 2011, 132, 402–408. doi:10.1016/j.jfluchem.2011.03.023 |
| 6. | Frederickson, M. Tetrahedron 1997, 53, 403–425. doi:10.1016/S0040-4020(96)01095-2 |
| 32. | Bailey, W. J.; Cummins, E. W. J. Am. Chem. Soc. 1954, 76, 1932–1936. doi:10.1021/ja01636a058 |
| 4. | Gothelf, K. V.; Jørgensen, K. A. Chem. Rev. 1998, 98, 863–910. doi:10.1021/cr970324e |
| 5. | Padwa, A. 1,3-Dipolar Cycloadditions; John Wiley: New York, NY, 1984. |
| 33. | Xin, Y.; Zhao, J.; Gu, J.; Zhu, S. J. Fluorine Chem. 2011, 132, 402–408. doi:10.1016/j.jfluchem.2011.03.023 |
| 26. | Yarmolchuk, V. S.; Mukan, I. L.; Grygorenko, O. O.; Tolmachev, A. A.; Shishkina, S. V.; Shishkin, O. V.; Komarov, I. V. J. Org. Chem. 2011, 76, 7010–7016. doi:10.1021/jo200878t |
| 30. | Brant, M. G.; Bromba, C. M.; Wulff, J. E. J. Org. Chem. 2010, 75, 6312–6315. doi:10.1021/jo101389c |
| 40. | Traynelis, V. J.; Hergenrother, W. L. J. Org. Chem. 1964, 29, 221–222. doi:10.1021/jo01024a505 |
| 22. | Barr, D. A.; Donegan, G.; Grigg, R. J. Chem. Soc., Perkin Trans. 1 1989, 1550–1551. doi:10.1039/P19890001550 |
| 23. | Jones, R. C. F.; Rafiq, S.; Elsegood, M. R. J.; McKee, V.; Slater, M. J. Chem.–Asian J. 2010, 5, 461–465. doi:10.1002/asia.200900547 |
| 24. | Lakshmi, N. V.; Thirumurugan, P.; Jayakumar, C.; Perumal, P. T. Synlett 2010, 955–961. doi:10.1055/s-0029-1219550 |
| 25. | Atienza, R. L.; Roth, H. S.; Scheidt, K. A. Chem. Sci. 2011, 2, 1772–1776. doi:10.1039/c1sc00194a |
| 31. | Brant, M. G.; Wulff, J. E. Org. Lett. 2012, 14, 5876–5879. doi:10.1021/ol3027939 |
| 13. | Clark, R. B.; Pearson, W. H. Org. Lett. 1999, 1, 349–352. doi:10.1021/ol990677v |
| 14. | Huscroft, I. T.; Carlson, E. J.; Chicchi, G. G.; Kurtz, M. M.; London, C.; Raubo, P.; Wheeldon, A.; Kulagowski, J. J. Bioorg. Med. Chem. Lett. 2006, 16, 2008–2012. doi:10.1016/j.bmcl.2005.12.069 |
| 15. | Peese, K. M.; Gin, D. Y. Chem.–Eur. J. 2008, 14, 1654–1665. doi:10.1002/chem.200701290 |
| 16. | Kudryavtstev, K. V.; Ivantcova, P. M.; Churakov, A. V.; Vasin, V. A. Tetrahedron Lett. 2012, 53, 4300–4303. doi:10.1016/j.tetlet.2012.05.160 |
| 17. | Llamas, T.; Arrayás, R. G.; Carretero, J. C. Org. Lett. 2006, 8, 1795–1798. doi:10.1021/ol060314c |
| 18. | Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Chem.–Eur. J. 2011, 17, 14224–14233. doi:10.1002/chem.201101606 |
| 19. | Tong, M.-C.; Li, J.; Tao, H.-Y.; Li, Y.-X.; Wang, C.-J. Chem.–Eur. J. 2011, 17, 12922–12927. doi:10.1002/chem.201102430 |
| 20. | Yamashita, Y.; Imaizumi, T.; Kobayashi, S. Angew. Chem., Int. Ed. 2011, 50, 4893–4896. doi:10.1002/anie.201008272 |
| 21. | González-Esguevillas, M.; Adrio, J.; Carretero, J. C. Chem. Commun. 2012, 48, 2149–2151. doi:10.1039/c2cc17149j |
| 38. | All calculations were performed at the B3LYP/6-31G* level and were done using the Spartan 06 Molecular Modeling package. |
| 10. | Huisgen, R. J. Org. Chem. 1976, 41, 403–419. doi:10.1021/jo00865a001 |
| 11. | Zhu, X.-F.; Henry, C. E.; Kwon, O. Tetrahedron 2005, 61, 6276–6282. doi:10.1016/j.tet.2005.03.104 |
| 12. | Partridge, K. M.; Guzei, I. A.; Yoon, T. P. Angew. Chem., Int. Ed. 2010, 49, 930–934. doi:10.1002/anie.200905801 |
| 27. | Müller, C.; Schweig, A. Tetrahedron 1973, 29, 3973–3977. doi:10.1016/0040-4020(73)80222-4 |
| 28. | Caramella, P.; Albini, E.; Bandiera, T.; Coda, A. C.; Grünanger, P.; Albini, F. M. Tetrahedron 1983, 39, 689–699. doi:10.1016/S0040-4020(01)91846-0 |
| 29. | Barzaghi, M.; Beltrame, P. L.; Croce, P. D.; Buttero, P. D.; Licandro, E.; Maiorana, S.; Zecchi, G. J. Org. Chem. 1983, 48, 3807–3810. doi:10.1021/jo00169a041 |
| 39. | Chou, T.-s.; Chang, L.-J.; Tso, H.-H. J. Chem. Soc., Perkin Trans. 1 1986, 1039–1042. doi:10.1039/P19860001039 |
© 2013 Wong et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)