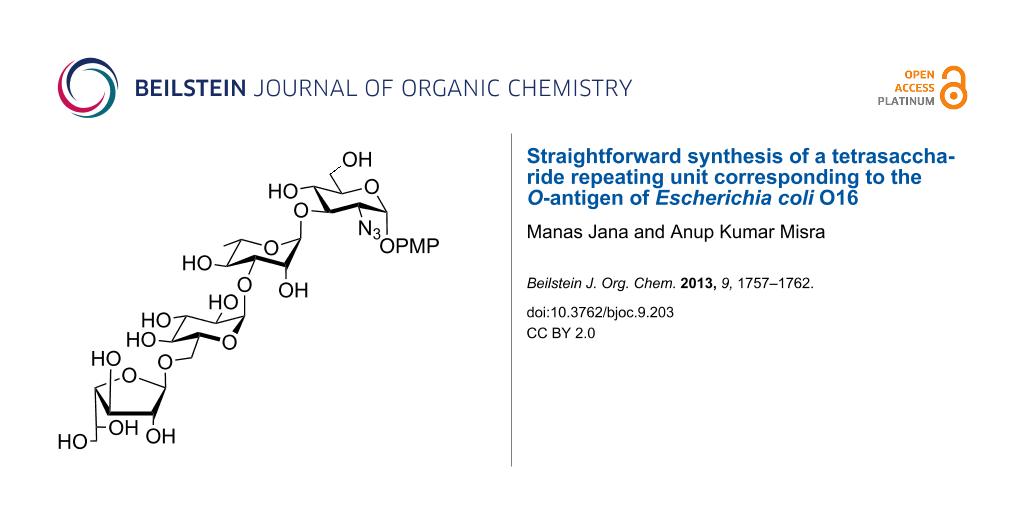
Abstract
A straightforward synthesis of the tetrasaccharide repeating unit of the O-antigen of Escherichia coli O16 has been achieved following a sequential glycosylation strategy. A minimum number of steps was used for the synthesis of the target compound involving a one-pot glycosylation and a protecting group manipulation. All intermediate reactions afford their products in high yield, and the glycosylation steps are stereoselective.

Graphical Abstract
Introduction
Neonatal meningitis is a serious concern in developing countries [1]. The symptoms associated with this disease are unspecific and may ultimately lead to sepsis [2]. The common cause of the neonatal meningitis are bacterial infections in blood, and they start with the bacteria colonizing the gastrointestinal tract [3,4]. Microorganisms associated with neonatal meningitis are Streptococcus, Escherichia coli (E. coli) and Listeria monocytogenes [5,6]. Major E. coli strains causing neonatal meningitis are O1, O6, O7, O16, O18 and O83 [7]. Like many other E. coli strains, meningitis causing E. coli O16 is encapsulated and exhibits the K1 polysaccharides [8]. The structure of the E. coli O16 polysaccharide has been established by Jann et al. [9], which is a tetrasaccharide repeating unit containing D-glucosamine, L-rhamnose, D-glucose and D-galactofuranose moieties in a 1:1:1:1 ratio (Figure 1).
![[1860-5397-9-203-1]](/bjoc/content/figures/1860-5397-9-203-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of the tetrasaccharide repeating unit of the O-antigen of Escherichia coli O16.
Figure 1: Structure of the tetrasaccharide repeating unit of the O-antigen of Escherichia coli O16.
The emergence of multi drug resistant bacterial strains forces medicinal chemists to develop new approaches to combat bacterial infections. Since the structure of the O-antigen influences the virulence property of the pathogen, several reports appeared in the past on the development of glycoconjugate based therapeutics against bacterial infections [10-12]. Detailed biological studies of the glycoconjugates require a significant quantity of the oligosaccharides, which is difficult to isolate from natural sources. Hence, the development of chemical synthetic strategies for the synthesis of oligosaccharides is essential. In this context, a straightforward synthesis of the tetrasaccharide corresponding to the O-antigen of E. coli O16 as its p-methoxyphenyl glycoside has been developed and is presented herein (Figure 2).
![[1860-5397-9-203-2]](/bjoc/content/figures/1860-5397-9-203-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of the synthesized pentasaccharide and its synthetic intermediates.
Figure 2: Structure of the synthesized pentasaccharide and its synthetic intermediates.
Results and Discussion
The target tetrasaccharide as its p-methoxyphenyl (PMP) glycoside was synthesized following a sequential glycosylation approach from the suitably functionalized monosaccharide intermediates 2 [13], 3 [14], 4 [15] and 5 [16]. These monosaccharide intermediates were prepared from the commercially available reducing sugars by using a number of recently developed reaction conditions (Figure 2). The notable features of the synthetic strategy include, (a) the use of thioglycosides as glycosyl donors in all glycosylation reactions; (b) the application of iodonium ion mediated glycosylation conditions; (c) the use of p-methoxybenzyl (PMB) ether protection as an in situ removable protecting group in a one-pot glycosylation reaction and its removal [17] and (d) the use of galactofuranosidic thioglycoside as a glycosyl donor.
The iodonium ion promoted stereoselective glycosylation of compounds 2 and 3 in the presence of a combination of N-iodosuccinimide (NIS) and triflic acid (TfOH) [18,19], followed by the removal of the p-methoxybenzyl (PMB) group in a one-pot reaction [17] by tuning the reaction conditions furnished the disaccharide acceptor 6 in 76% yield. The PMB group acted as an in situ temporary protecting group, which was removed after the glycosylation took place in the same pot. Formation of compound 6 was supported by its spectral analysis (signals at δ 5.46 (d, J = 3.0 Hz, H-1A), 5.09 (br s, 1H, H-1B) and at δ 98.1 (C-1A), 97.8 (C-1B) in the 1H and 13C NMR spectra respectively). The coupling of compound 6 with thioglycoside 4 in the presence of a combination of NIS and TfOH [18,19] in CH2Cl2/Et2O (1:3, v/v) furnished the 1,2-cis glycosylated compound 7 in 73% yield together with a minor quantity (~8%) of its other isomer, which was separated by chromatography. Spectral analysis of compound 7 confirmed its formation (signals at δ 5.46 (d, J = 3.5 Hz, H-1A), 5.11 (d, J = 3.5 Hz, H-1C), 5.06 (br s, H-1B) and at δ 98.1 (C-1A), 98.0 (C-1B), 92.5 (C-1C) in the 1H and 13C NMR spectra, respectively). The de-O-acetylation of compound 7 by using sodium methoxide furnished the trisaccharide acceptor 8 in 94% yield. The stereoselective glycosylation of compound 8 with D-galactofuranosyl thioglycoside 5 by using a combination of NIS/TfOH furnished the tetrasaccharide derivative 9 in 72% yield. The formation of compound 9 was supported by its spectral analysis (signals at δ 5.47 (d, J = 3.5 Hz, H-1A), 5.17 (br s, H-1B), 4.87 (d, J = 3.0 Hz, H-1C), 4.80 (br s, H-1D) and at δ 105.6 (C-1D), 99.9 (C-1B), 97.9 (C-1A), 93.8 (C-1C) in the 1H and 13C NMR spectra respectively). Compound 9 was subjected to a series of reactions involving (a) a catalytic transfer hydrogenation with triethylsilane and 10% Pd/C [20]; (b) an acetylation using acetic anhydride and pyridine, and (c) a saponification reaction with sodium methoxide to furnish compound 1, which was purified over a Sephadex® LH-20 gel to give the pure compound 1 in 64% overall yield. The structure of compound 1 was unambiguously confirmed by spectral analysis (signals at δ 4.89 (br s, H-1D), 4.88 (d, J = 3.0 Hz, H-1A), 4.84 (d, J = 3.0 Hz, H-1C), 4.76 (br s, H-1B) and at δ 107.6 (C-1D), 100.5 (C-1B), 95.9 (C-1A), 95.0 (C-1C) in the 1H and 13C NMR spectra respectively) (Scheme 1).
![[1860-5397-9-203-i1]](/bjoc/content/inline/1860-5397-9-203-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents: (a) N-iodosuccinimide (NIS), TfOH, CH2Cl2, MS 4 Å, −30 °C, 1 h, then 0 °C, 1 h, 76%; (b) NIS, TfOH, CH2Cl2/Et2O (1:3, v/v), MS 4 Å, −40 °C, 1 h, 73%; (c) 0.1 M CH3ONa, CH3OH, room temperature, 2 h, 94%; (d) NIS, TfOH, CH2Cl2, MS 4 Å, −20 °C, 1 h, 72%; (e) Et3SiH, 10% Pd/C, CH3OH, AcOH, room temperature, 12 h; (f) acetic anhydride, pyridine, room temperature, 1 h; (g) 0.1 M CH3ONa, CH3OH, room temperature, 2 h, 64% in three steps.
Scheme 1: Reagents: (a) N-iodosuccinimide (NIS), TfOH, CH2Cl2, MS 4 Å, −30 °C, 1 h, then 0 °C, 1 h, 76%; (b) ...
Conclusion
In summary, a straightforward synthetic strategy was developed for the synthesis of the tetrasaccharide 1 as its p-methoxyphenyl glycoside corresponding to the O-antigen of E. coli O16. The target compound was synthesized by using a minimum number of steps and by applying recently developed elegant synthetic methodologies. Both the yields of the protecting group manipulations and the stereoselectivity of the glycosylation reactions were excellent.
Experimental
General methods are similar as described in an earlier report [21].
p-Methoxyphenyl (2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1→3)-2-azido-4,6-O-benzylidene-2-deoxy-α-D-glucopyranoside (6): Similar as described in [21]. To a solution of compounds 2 (2 g, 5.00 mmol) and 3 (2.5 g, 5.42 mmol) in anhydrous CH2Cl2 (10 mL) was added MS 4 Å (2 g), and the reaction mixture was stirred under an argon atmosphere at room temperature for 30 min. The reaction mixture was cooled to −30 °C. To the cooled reaction mixture N-iodosuccinimide (NIS; 1.3 g, 5.77 mmol) and trifluoromethanesulfonic acid (TfOH; 50 μL) were added, and the mixture was stirred at the same temperature for 1 h. The temperature of the reaction mixture was raised to 0 °C and it was stirred at 0 °C for another 1 h. The reaction mixture was filtered through a bed of Celite® and washed with CH2Cl2 (100 mL). The combined organic layers were successively washed with 5% Na2S2O3, satd. NaHCO3, and water, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 by using hexane/EtOAc (4:1) as an eluant to give the pure compound 6 (2.6 g, 76%). Yellow oil; [α]D25 +77 (c 1.5, CHCl3); IR (neat): 3468, 2931, 2869, 2111, 1664, 1498, 1439, 1411, 1389, 1255, 1098, 1063, 1063, 1028, 661 cm−1; 1H NMR (CDCl3, 500 MHz) δ 7.45–7.23 (m, 10H, Ar-H), 7.01 (d, J = 9.0 Hz, 2H, Ar-H), 6.81 (d, J = 9.0 Hz, 2H, Ar-H), 5.50 (s, 1H, PhCH), 5.46 (d, J = 3.0 Hz, 1H, H-1A), 5.20–5.19 (m, 1H, H-2B), 5.09 (br s, 1H, H-1B), 4.72 (d, J = 11.5 Hz, 1H, PhCH2), 4.59 (d, J = 11.5 Hz, 1H, PhCH2), 4.30 (t, J = 9.5 Hz each, 1H, H-3A), 4.26–4.23 (m, 1H, H-6aA), 4.12–4.05 (m, 2H, H-3B, H-5A), 4.02–3.88 (m, 1H, H-5B), 3.78 (s, 3H, OCH3), 3.73–3.69 (m, 1H, H-6bA), 3.58 (t, J = 10.0 Hz each, 1H, H-4A), 3.42 (dd, J = 10.0, 3.5 Hz, 1H, H-2A), 3.26 (t, J = 9.5 Hz each, 1H, H-4B), 2.16 (s, 3H, COCH3), 0.86 (d, J = 6.0 Hz, 3H, CCH3); 13C NMR (CDCl3, 125 MHz) δ 169.9, (3 COCH3), 155.6–114.7 (Ar-C), 102.1 (PhCH), 98.1 (C-1A), 97.8 (C-1B), 81.5 (C-4B), 80.1 (C-4A), 74.8 (PhCH2), 73.7 (C-3A), 72.7 (C-2B), 69.9 (C-3B), 68.7 (C-6A), 67.7 (C-5B), 64.1 (C-5A), 63.5 (C-2A), 55.5 (OCH3), 21.0 (COCH3), 17.3 (CCH3); ESI–MS: 700.2 [M + Na]+; Anal. calcd for C35H39N3O11: C, 62.03; H, 5.80%; found: C, 61.86; H, 6.00%.
p-Methoxyphenyl (6-O-acetyl-2,3,4-tri-O-benzyl-α-D-glucopyranosyl)-(1→3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1→3)-2-azido-4,6-O-benzylidene-2-deoxy-α-D-glucopyranoside (7): Similar as described in [21]. To a solution of compounds 6 (2 g, 2.95 mmol) and 4 (1.7 g, 3.16 mmol) in anhydrous CH2Cl2/Et2O (10 mL; 1:3, v/v) was added MS 4 Å (2 g), and the reaction mixture was stirred under an argon atmosphere at room temperature for 30 min. The reaction mixture was cooled to −40 °C. To the cooled reaction mixture NIS (780 mg, 3.46 mmol) and TfOH (15 μL) were added, and the mixture was stirred at same temperature for 1 h. The reaction mixture was filtered through a bed of Celite® and washed with CH2Cl2 (100 mL). The combined organic layers were successively washed with 5% Na2S2O3, satd. NaHCO3, and water, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 by using hexane/EtOAc (3:1) as an eluant to give the pure compound 7 (2.5 g, 73%). Yellow oil; [α]D25 +67 (c 1.5, CHCl3); IR (neat): 3469, 2931, 2867, 2108, 1745, 1671, 1507, 1439, 1388, 1255, 1095, 1028, 865, 832, 753, 701, 660 cm−1; 1H NMR (CDCl3, 500 MHz) δ 7.43–7.09 (m, 25H, Ar-H), 7.01 (d, J = 9.0 Hz, 2H, Ar-H), 6.82 (d, J = 9.0 Hz, 2H, Ar-H), 5.53 (s, 1H, PhCH), 5.46 (d, J = 3.5 Hz, 1H, H-1A), 5.41–5.40 (m, 1H, H-2B), 5.11 (d, J = 3.5 Hz, 1H, H-1C), 5.06 (br s, 1H, H-1B), 5.03 (d, J = 11.0 Hz, 1H, PhCH2), 4.85 (d, J = 11.0 Hz, 1H, PhCH2), 4.84–4.80 (m, 2H, PhCH2), 4.68 (d, J = 12.0 Hz, 1H, PhCH2), 4.62 (d, J = 11.0 Hz, 1H, PhCH2), 4.52 (d, J = 10.5 Hz, 1H, PhCH2), 4.48 (d, J = 10.5 Hz, 1H, PhCH2), 4.26 (t, J = 9.5 Hz each, 1H, H-3A), 4.25–4.23 (m, 1H, H-6aA), 4.11 (dd, J = 10.0, 3.0 Hz, 1H, H-3A), 4.10–4.03 (m, 4H, H-5A, H-5C, H-6aC, H-6bA), 4.02–3.99 (m, 1H, H-5B), 3.93–3.90 (m, 1H, H-6bC), 3.77 (s, 3H, OCH3), 3.74–3.70 (m, 1H, H-4C), 3.65 (t, J = 9.0 Hz each, 1H, H-4A), 3.54 (dd, J = 10.0, 3.5 Hz, 1H, H-2C), 3.51 (t, J = 9.0 Hz each, 1H, H-3C), 3.45 (t, J = 10.0 Hz each, 1H, H-4B), 3.42 (dd, J = 10.0, 3.0 Hz, 1H, H-2A), 1.95, 1.89 (2 s, 6H, 2COCH3), 0.90 (d, J = 6.0 Hz, 3H, CCH3); 13C NMR (CDCl3, 125 MHz) δ 170.4, 170.3 (2 COCH3), 155.8–114.7 (Ar-C), 102.0 (PhCH), 98.1 (C-1A), 98.0 (C-1B), 92.5 (C-1C), 82.0 (C-4C), 79.9 (C-4A), 79.4 (2 C, C-3B, C-4B), 76.7 (C-3C), 75.8 (PhCH2), 75.6 (PhCH2), 74.9 (PhCH2), 73.9 (C-2C), 72.1 (C-3A), 68.7 (C-6A), 68.5 (C-5B), 68.4 (C-5C), 67.6 (C-2B), 64.0 (C-5A), 63.6 (C-2A), 62.4 (C-6C), 55.5 (OCH3), 20.8, 20.7 (2 COCH3), 17.3 (CCH3); ESI–MS: 1174.4 [M + Na]+; Anal. calcd for C64H69N3O17: C, 66.71; H, 6.04%; found: C, 66.54; H, 6.20%.
p-Methoxyphenyl (2,3,4-tri-O-benzyl-α-D-glucopyranosyl)-(1→3)-(4-O-benzyl-α-L-rhamnopyranosyl)-(1→3)-2-azido-4,6-O-benzylidene-2-deoxy-α-D-glucopyranoside (8): Similar as described in [21]. A solution of compound 7 (2 g, 1.73 mmol) in 0.1 M CH3ONa in CH3OH (20 mL) was stirred at room temperature for 2 h. The reaction mixture was neutralized with Dowex 50W-X8 (H+) resin, filtered and concentrated. The crude product was passed through a small pad of SiO2 by using hexane/EtOAc (1:1) as an eluant to give the pure compound 8 (1.8 g, 94%). Yellow oil; [α]D25 +57 (c 1.5, CHCl3); IR (neat): 3458, 3002, 2932, 2871, 2472, 2109, 1747, 1667, 1505, 1439, 1411, 1389, 1255, 1097, 1063, 865, 754, 700, 663 cm−1; 1H NMR (CDCl3, 500 MHz) δ 7.46–7.17 (m, 25H, Ar-H), 7.02 (d, J = 9.0 Hz, 2H, Ar-H), 6.84 (d, J = 9.0 Hz, 2H, Ar-H), 5.50 (s, 1H, PhCH), 5.47 (d, J = 3.5 Hz, 1H, H-1A), 5.18 (br s, 1H, H-1B), 4.94–4.85 (m, 3H, PhCH2), 4.83 (d, J = 3.5 Hz, 1H, H-1C), 4.80 (d, J = 11.5 Hz, 1H, PhCH2), 4.69–4.56 (m, 4H, PhCH2), 4.36 (t, J = 9.5 Hz each, 1H, H-3A), 4.27–4.22 (m, 1H, H-6aA), 4.17–3.96 (m, 5H, H-2B, H-5A, H-5B, H-6bA, H-6aC), 3.92 (dd, J = 10.0, 3.5 Hz, 1H, H-3B), 3.84–3.80 (m, 1H, H-6bC), 3.78 (s, 3H, OCH3), 3.69 (t, J = 10.0 Hz each, 1H, H-4A), 3.58 (t, J = 9.5 Hz each, 1H, H-3C), 3.51 (t, J = 9.5 Hz each, 1H, H-4C), 3.49 (dd, J = 10.0, 3.5 Hz, 1H, H-2C), 3.48–3.40 (m, 3H, H-2A, H-4B, H-5C), 0.85 (d, J = 6.0 Hz, 3H, CCH3); 13C NMR (CDCl3, 125 MHz) δ 155.6–114.7 (Ar-C), 102.2 (PhCH), 99.8 (C-1A), 97.9 (C-1B), 94.0 (C-1C), 82.2 (C-4C), 80.2 (C-4A), 78.9 (2 C, C-3B, C-4B), 77.4 (C-3C), 76.7 (C-2C), 75.6 (PhCH2), 75.2 (PhCH2), 74.9 (PhCH2), 74.3 (PhCH2), 74.0 (C-3A), 71.5 (C-5C), 68.7 (C-5B), 67.5 (C-2B), 64.3 (C-5A), 63.6 (C-2A), 61.2 (C-6C), 55.5 (OCH3), 17.2 (CCH3); ESI–MS: 1132.4 [M + Na]+; Anal. calcd for C62H67N3O16: C, 67.07; H, 6.08%; found: C, 66.88; H, 6.30%.
p-Methoxyphenyl (2,3,5,6-tetra-O-acetyl-β-D-galactofuranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-glucopyranosyl)-(1→3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1→3)-2-azido-4,6-O-benzylidene-2-deoxy-α-D-glucopyranoside (9): Similar as described in [21]. To a solution of compounds 8 (1.5 g, 1.35 mmol) and 5 (580 mg, 1.48 mmol) in anhydrous CH2Cl2 (5 mL) was added MS 4 Å (1 g), and the reaction mixture was stirred under an argon atmosphere at room temperature for 30 min. The reaction mixture was cooled to −20 °C. To the cooled reaction mixture NIS (350 mg, 1.56 mmol) and TfOH (5 μL) were added, and the mixture was stirred at the same temperature for an additional hour. The reaction mixture was filtered through a bed of Celite® and washed with CH2Cl2 (50 mL). The combined organic layers were successively washed with 5% Na2S2O3, satd. NaHCO3, and water, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 by using hexane/EtOAc (5:1) as an eluant to give the pure compound 9 (1.4 g, 72%). Yellow oil; [α]D25 +28 (c 1.5, CHCl3); IR (neat): 3469, 2931, 2867, 2108, 1745, 1671, 1507, 1439, 1388, 1255, 1095, 1028, 865, 753, 701, 660 cm−1; 1H NMR (CDCl3, 500 MHz) δ 7.46–7.14 (m, 25H, Ar-H), 7.03 (d, J = 9.0 Hz, 2H, Ar-H), 6.84 (d, J = 9.0 Hz, 2H, Ar-H), 5.52 (s, 1H, PhCH), 5.47 (d, J = 3.5 Hz, 1H, H-1A), 5.32–5.28 (m, 1H, H-5D), 5.17 (br s, 1H, H-1B), 5.00 (d, J = 2.0 Hz, 1H, H-2D), 4.95–4.89 (m, 3H, PhCH2), 4.87 (d, J = 3.0 Hz, 1H, H-1C), 4.82 (d, J = 11.0 Hz, 1H, PhCH2), 4.80 (br s, 1H, H-1D), 4.68 (d, J = 11.5 Hz, 1H, PhCH2), 4.57 (d, J = 11.5 Hz, 1H, PhCH2), 4.55 (d, J = 11.5 Hz, 1H, PhCH2), 4.53 (d, J = 11.5 Hz, 1H, PhCH2), 4.35 (t, J = 9.5 Hz each, 1H, H-3A), 4.27–4.22 (m, 2H, H-3D, H-6aC), 4.20–4.17 (m, 1H, H-4D), 4.12–4.07 (m, 3H, H-5A, H-6aA, H-6bC), 4.06–4.05 (m, 1H, H-2B), 4.02 (t, J = 9.5 Hz each, 1H, H-3C), 4.00–3.98 (m, 1H, H-5B), 3.93 (dd, J = 10.0, 3.5 Hz, 1H, H-3B), 3.89–3.85 (m, 1H, H-5C), 3.78 (s, 3H, OCH3), 3.75–3.72 (m, 1H, H-6bA), 3.65 (t, J = 9.5 Hz each, 1H, H-4C), 3.59 (t, J = 9.5 Hz each, 1H, H-4A), 3.52–3.49 (m, 1H, H-6aD), 3.46 (dd, J = 10.0, 3.0 Hz, 1H, H-2C), 3.43 (dd, J = 10.0, 3.0 Hz, 1H, H-2A), 3.40 (t, J = 10.0 Hz each, 1H, H-4B), 3.32–3.28 (m, 1H, H-6bD), 2.10, 2.04, 2.03, 1.97 (4 s, 12H, 4 COCH3), 0.85 (d, J = 6.0 Hz, 3H, CCH3); 13C NMR (CDCl3, 125 MHz) δ 171.2, 170.8, 170.7, 170.6 (4 COCH3), 155.8–114.7 (Ar-C), 105.6 (C-1D), 102.2 (PhCH), 99.9 (C-1B), 97.9 (C-1A), 93.8 (C-1C), 82.2 (C-3C), 81.2 (C-2D), 80.2 (C-4A), 79.4 (C-4D), 79.0 (C-2C), 78.9 (C-4B), 77.4 (C-4C), 76.4 (C-3B), 76.1 (C-2B), 75.6 (PhCH2), 75.2 (PhCH2), 74.8 (PhCH2), 74.2 (PhCH2), 74.1 (C-3A), 70.1 (C-5C), 68.9 (C-5D), 68.7 (C-6A), 67.6 (C-3D), 67.4 (C-5B), 66.0 (C-6D), 64.2 (C-5A), 63.4 (C-2A), 62.8 (C-6C), 55.6 (OCH3), 20.8 (2 C), 20.7 (2 C) (4 COCH3), 18.1 (CCH3); MALDI–MS: 1462.5 [M + Na]+; Anal. calcd for C76H85N3O25: C, 63.37; H, 5.95%; found: C, 63.20; H, 6.18%.
p-Methoxyphenyl (β-D-galactofuranosyl)-(1→6)-(α-D-glucopyranosyl)-(1→3)-(α-L-rhamnopyranosyl)-(1→3)-2-acetamido-2-deoxy-α-D-glucopyranoside (1): Similar as described in [21]. To a solution of compound 9 (1 g, 0.69 mmol) in CH3OH/AcOH (10 mL, 20:1, v/v), were added 10% Pd/C (100 mg) and Et3SiH (2 mL, 12.5 mmol), and the reaction mixture was stirred at room temperature for 12 h. The reaction mixture was filtered through a bed of Celite®, washed with warm CH3OH, and concentrated under reduced pressure. A solution of the crude product in acetic anhydride/pyridine (2 mL, 1:1 v/v) was kept at room temperature for 1 h and concentrated under reduced pressure. A solution of the acetylated crude product in 0.1 M CH3ONa in CH3OH (5 mL) was stirred at room temperature for 2 h. The reaction mixture was neutralized with Dowex 50W-X8 (H+) resin, filtered, and concentrated. The crude product was passed through a Sephadex® LH-20 column by using CH3OH/H2O (2:1) as an eluant to give the pure compound 1 (345 mg, 64%). Glass; [α]D25 −6 (c 1.5, H2O); IR (KBr): 3466, 2945, 1632, 1376, 1165, 1067, 697 cm−1; 1H NMR (D2O, 500 MHz) δ 7.01 (d, J = 9.0 Hz, 2H, Ar-H), 6.87 (d, J = 9.0 Hz, 2H, Ar-H), 4.89 (br s, 1H, H-1D), 4.88 (d, J = 3.0 Hz, 1H, H-1A), 4.84 (d, J = 3.0 Hz, 1H, H-1C), 4.76 (br s, 1H, H-1B), 4.20–3.98 (m, 2H, H-2D, H-4D), 3.95–3.89 (m, 3H, H-2A, H-3D, H-5B), 3.88–3.80 (m, 3H, H-3B, H-5C, H-6aC), 3.76–3.68 (m, 5H, H-3C, H-4A, H-5A, H-6abD), 3.65 (s, 3H, OCH3), 3.64–3.62 (m, 2H, H-4C, H-5D), 3.61–3.56 (m, 2H, H-6aA, H-6bC), 3.55–3.50 (m, 2H, H-3A, H-6bA), 3.48–3.39 (m, 3H, H-2B, H-2C, H-4B), 1.94 (s, 3H, COCH3), 1.12 (d, J = 6.0 Hz, 3H, CCH3); 13C NMR (D2O, 125 MHz) δ 174.0 (COCH3), 155.4–115.0 (Ar-C), 107.6 (C-1D), 100.5 (C-1B), 95.9 (C-1A), 95.0 (C-1C), 82.6 (C-3D), 80.8 (C-4D), 76.3 (C-5B), 72.8 (C-3C), 72.1 (C-4C), 71.5 (2 C, C-2D, C-3A), 70.7 (C-3B), 70.5 (C-2B), 70.1 (C-4B), 69.2 (2 C, C-2C, C-4A), 68.8 (C-5D), 68.3 (C-5C), 67.6 (C-5A), 66.1 (C-6C), 62.7 (C-6D), 60.5 (C-6A), 55.0 (OCH3), 54.9 (C-2A), 21.8 (COCH3), 16.5 (CCH3); ESI–MS: 804.2 [M + Na]+; Anal. calcd for C31H47N3O20: C, 47.63; H, 6.06%; found: C, 47.46; H, 6.22%.
Supporting Information
| Supporting Information File 1: 1D and 2D NMR spectra of compounds 1 and 6–9. | ||
| Format: PDF | Size: 3.0 MB | Download |
References
-
Osrin, D.; Vergnano, S.; Costello, A. Curr. Opin. Infect. Dis. 2004, 17, 217–224. doi:10.1097/00001432-200406000-00008
Return to citation in text: [1] -
Moreno, M. T.; Vargas, S.; Poveda, R.; Sáez-Llorens, X. Pediatr. Infect. Dis. J. 1994, 13, 516–520. doi:10.1097/00006454-199406000-00010
Return to citation in text: [1] -
De Louvois, J. J. Antimicrob. Chemother. 1994, 34 (Suppl. A), 61–73. doi:10.1093/jac/34.suppl_A.61
Return to citation in text: [1] -
Kavuncuoğlu, S.; Gürsoy, S.; Türel, Ö.; Aldemir, E. Y.; Hoşaf, E. J. Infect. Dev. Countries 2013, 7, 73–81. doi:10.3855/jidc.2652
Return to citation in text: [1] -
Zaidi, A. K. M.; Thaver, D.; Ali, S. A.; Khan, T. A. Pediatr. Infect. Dis. J. 2009, 28, S10–S18. doi:10.1097/INF.0b013e3181958769
Return to citation in text: [1] -
Mulder, C. J. J.; Zanen, H. C. Eur. J. Pediatr. 1986, 145, 60–62. doi:10.1007/BF00441855
Return to citation in text: [1] -
Ørskov, I.; Ørskov, F.; Jann, B.; Jann, K. Bacteriol. Rev. 1977, 41, 667–710.
Return to citation in text: [1] -
Ørskov, F.; Ørskov, I.; Sutton, A.; Schneerson, R.; Lin, W.; Egan, W.; Hoff, G. E.; Robbins, J. B. J. Exp. Med. 1979, 149, 669–685. doi:10.1084/jem.149.3.669
Return to citation in text: [1] -
Jann, B.; Shashkov, A. S.; Kochanowski, H.; Jann, K. Carbohydr. Res. 1994, 264, 305–311. doi:10.1016/S0008-6215(05)80014-X
Return to citation in text: [1] -
Kuberan, B.; Linhardt, R. J. Curr. Org. Chem. 2000, 4, 653–677. doi:10.2174/1385272003376111
Return to citation in text: [1] -
Doores, K. J.; Gamblin, D. P.; Davis, B. G. Chem.–Eur. J. 2006, 12, 656–665. doi:10.1002/chem.200500557
Return to citation in text: [1] -
Osborn, H. M. I.; Evans, P. G.; Gemmell, N.; Osborne, S. D. J. Pharm. Pharmacol. 2004, 56, 691–702. doi:10.1211/0022357023619
Return to citation in text: [1] -
Sau, A.; Panchadhayee, R.; Ghosh, D.; Misra, A. K. Carbohydr. Res. 2012, 352, 18–22. doi:10.1016/j.carres.2012.01.026
Return to citation in text: [1] -
Mukherjee, C.; Misra, A. K. Glycoconjugate J. 2008, 25, 611–624. doi:10.1007/s10719-008-9107-y
Return to citation in text: [1] -
Misra, A. K.; Roy, N. Carbohydr. Res. 1995, 278, 103–111. doi:10.1016/0008-6215(95)00231-X
Return to citation in text: [1] -
Tiwari, P.; Misra, A. K. Glycoconjugate J. 2008, 25, 85–99. doi:10.1007/s10719-007-9056-x
Return to citation in text: [1] -
Bhattacharyya, S.; Magnusson, B. G.; Wellmar, U.; Nilsson, U. J. J. Chem. Soc., Perkin Trans. 1 2001, 886–890. doi:10.1039/B009448J
Return to citation in text: [1] [2] -
Veeneman, G. H.; van Leeuwen, S. H.; van Boom, J. H. Tetrahedron Lett. 1990, 31, 1331–1334. doi:10.1016/S0040-4039(00)88799-7
Return to citation in text: [1] [2] -
Konradsson, P.; Udodong, U. E.; Fraser-Reid, B. Tetrahedron Lett. 1990, 31, 4313–4316. doi:10.1016/S0040-4039(00)97609-3
Return to citation in text: [1] [2] -
Santra, A.; Ghosh, T.; Misra, A. K. Beilstein J. Org. Chem. 2013, 9, 74–78. doi:10.3762/bjoc.9.9
Return to citation in text: [1] -
Sau, A.; Misra, A. K. PLoS One 2012, 7, e37291. doi:10.1371/journal.pone.0037291
Return to citation in text: [1] [2] [3] [4] [5] [6]
| 21. | Sau, A.; Misra, A. K. PLoS One 2012, 7, e37291. doi:10.1371/journal.pone.0037291 |
| 20. | Santra, A.; Ghosh, T.; Misra, A. K. Beilstein J. Org. Chem. 2013, 9, 74–78. doi:10.3762/bjoc.9.9 |
| 21. | Sau, A.; Misra, A. K. PLoS One 2012, 7, e37291. doi:10.1371/journal.pone.0037291 |
| 1. | Osrin, D.; Vergnano, S.; Costello, A. Curr. Opin. Infect. Dis. 2004, 17, 217–224. doi:10.1097/00001432-200406000-00008 |
| 17. | Bhattacharyya, S.; Magnusson, B. G.; Wellmar, U.; Nilsson, U. J. J. Chem. Soc., Perkin Trans. 1 2001, 886–890. doi:10.1039/B009448J |
| 5. | Zaidi, A. K. M.; Thaver, D.; Ali, S. A.; Khan, T. A. Pediatr. Infect. Dis. J. 2009, 28, S10–S18. doi:10.1097/INF.0b013e3181958769 |
| 6. | Mulder, C. J. J.; Zanen, H. C. Eur. J. Pediatr. 1986, 145, 60–62. doi:10.1007/BF00441855 |
| 18. | Veeneman, G. H.; van Leeuwen, S. H.; van Boom, J. H. Tetrahedron Lett. 1990, 31, 1331–1334. doi:10.1016/S0040-4039(00)88799-7 |
| 19. | Konradsson, P.; Udodong, U. E.; Fraser-Reid, B. Tetrahedron Lett. 1990, 31, 4313–4316. doi:10.1016/S0040-4039(00)97609-3 |
| 3. | De Louvois, J. J. Antimicrob. Chemother. 1994, 34 (Suppl. A), 61–73. doi:10.1093/jac/34.suppl_A.61 |
| 4. | Kavuncuoğlu, S.; Gürsoy, S.; Türel, Ö.; Aldemir, E. Y.; Hoşaf, E. J. Infect. Dev. Countries 2013, 7, 73–81. doi:10.3855/jidc.2652 |
| 17. | Bhattacharyya, S.; Magnusson, B. G.; Wellmar, U.; Nilsson, U. J. J. Chem. Soc., Perkin Trans. 1 2001, 886–890. doi:10.1039/B009448J |
| 2. | Moreno, M. T.; Vargas, S.; Poveda, R.; Sáez-Llorens, X. Pediatr. Infect. Dis. J. 1994, 13, 516–520. doi:10.1097/00006454-199406000-00010 |
| 18. | Veeneman, G. H.; van Leeuwen, S. H.; van Boom, J. H. Tetrahedron Lett. 1990, 31, 1331–1334. doi:10.1016/S0040-4039(00)88799-7 |
| 19. | Konradsson, P.; Udodong, U. E.; Fraser-Reid, B. Tetrahedron Lett. 1990, 31, 4313–4316. doi:10.1016/S0040-4039(00)97609-3 |
| 13. | Sau, A.; Panchadhayee, R.; Ghosh, D.; Misra, A. K. Carbohydr. Res. 2012, 352, 18–22. doi:10.1016/j.carres.2012.01.026 |
| 15. | Misra, A. K.; Roy, N. Carbohydr. Res. 1995, 278, 103–111. doi:10.1016/0008-6215(95)00231-X |
| 21. | Sau, A.; Misra, A. K. PLoS One 2012, 7, e37291. doi:10.1371/journal.pone.0037291 |
| 10. | Kuberan, B.; Linhardt, R. J. Curr. Org. Chem. 2000, 4, 653–677. doi:10.2174/1385272003376111 |
| 11. | Doores, K. J.; Gamblin, D. P.; Davis, B. G. Chem.–Eur. J. 2006, 12, 656–665. doi:10.1002/chem.200500557 |
| 12. | Osborn, H. M. I.; Evans, P. G.; Gemmell, N.; Osborne, S. D. J. Pharm. Pharmacol. 2004, 56, 691–702. doi:10.1211/0022357023619 |
| 16. | Tiwari, P.; Misra, A. K. Glycoconjugate J. 2008, 25, 85–99. doi:10.1007/s10719-007-9056-x |
| 21. | Sau, A.; Misra, A. K. PLoS One 2012, 7, e37291. doi:10.1371/journal.pone.0037291 |
| 9. | Jann, B.; Shashkov, A. S.; Kochanowski, H.; Jann, K. Carbohydr. Res. 1994, 264, 305–311. doi:10.1016/S0008-6215(05)80014-X |
| 21. | Sau, A.; Misra, A. K. PLoS One 2012, 7, e37291. doi:10.1371/journal.pone.0037291 |
| 8. | Ørskov, F.; Ørskov, I.; Sutton, A.; Schneerson, R.; Lin, W.; Egan, W.; Hoff, G. E.; Robbins, J. B. J. Exp. Med. 1979, 149, 669–685. doi:10.1084/jem.149.3.669 |
| 14. | Mukherjee, C.; Misra, A. K. Glycoconjugate J. 2008, 25, 611–624. doi:10.1007/s10719-008-9107-y |
| 21. | Sau, A.; Misra, A. K. PLoS One 2012, 7, e37291. doi:10.1371/journal.pone.0037291 |
© 2013 Jana and Misra; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)