Abstract
The efficiency of the intramolecular carbonickelation of substituted allylic ethers and amines has been studied to evaluate the influence of the groups borne by the double bond on this cyclization. The results show that when this reaction takes place, it affords only the 5-exo-trig cyclization products, viz. dihydrobenzofurans or indoles. Depending on the tethered heteroatom (O or N), the outcome of the cyclization differs. While allylic ethers are relatively poor substrates that undergo a side elimination and need an intracyclic double bond to proceed, allylic amines react well and afford indoline and indole derivatives. Finally, the synthesis of the trinuclear ACE core of a morphine-like skeleton was achieved by using NiBr2bipy catalysis.

Graphical Abstract
Introduction
Carbometalation is a reaction involving the addition of an organometallic species to a nonactivated alkene or alkyne to form a new carbon–carbon bond and generate a new organometallic entity, which may subsequently undergo synthetic transformations [1,2]. Even though these reactions have been known for over 85 years [3], they have emerged as practical organometallic tools only during the past forty years, in particular through the development of palladium chemistry [4]. The catalytic cycle starts with the oxidative addition of Pd(0) to generate a σ-arylpalladium(II), then a rapid insertion of a double or triple bond takes place [5]. This method was particularly applied in the “Mizoroki–Heck reaction” [6] for the synthesis of pharmaceutical and agrochemical intermediates using nonactivated olefins with high regio- and stereoselectivity [7]. Besides the typical intermolecular version, some intramolecular variants were developed leading to useful heterocycles [8-10]. Even if palladium is very efficient, nickel appears to be among the most promising metallic substitutes [11]. However, the tedious preparation of Ni(0) complexes such as Ni(cod)2 explains that nickel chemistry is hardly perceived as a realistic alternative to palladium, except in electrochemical processes [12,13]. Nevertheless, some nickel-catalyzed Heck vinylations have been recently reported on activated olefins [14,15]. Some years ago, we showed that the in situ generation of Ni(0) complexes in the presence of both the aromatic halide and the electrophile [16] represents an interesting alternative to electrochemical processes. The main advantages of the method are the use of an easily prepared Ni(II)bipy complex in combination with manganese dust as a reducing agent, which is not air sensitive, is compatible with fragile functions, and can be used in a catalytic amount. We showed that this nickel catalysis applies to cross-coupling reactions, efficiently leading to a variety of functionalized 2-arylpyridines [17]. More recently, this nickel-catalyzed reaction provided a convenient and mild method for a one-pot synthesis of substituted benzofurans, chromans and indoles by carbonickelation of alkynes [18]. We finally decided to extend the scope of this heterocyclization reaction to various nonactivated olefins in a nickel-catalyzed intramolecular base-free Heck-type coupling.
Results and Discussion
Scope of the reaction
We applied the Nickel-catalyzed intramolecular base-free Heck-type coupling to two model substrates, assumed to provide either a benzofuran or an indole core. Our first study involved the cyclization of allyl moieties such as allyl ether 1a or N-allyl protected anilines 1b–c, easily prepared by quantitative allylation of o-iodophenol and o-iodoanilines using allyl bromide in DMF (Scheme 1).
![[1860-5397-9-81-i1]](/bjoc/content/inline/1860-5397-9-81-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Following the protocol optimized for the carbonickelation of triple bonds [19], we exposed 1a–c to a mixture containing 0.2 equiv of NiBr2bipy and 2 equiv of finely grown manganese in DMF containing trace amounts of trifluoroacetic acid at 50 °C (Table 1). Disappointingly, using 1a the major new product recovered was the dimer 4a, obtained with trace amounts of several other byproducts (Table 1, entry 1). The formation of this symmetrical dimer suggests that: (i) the oxidative addition of Ni(0) into the carbon–iodine bond leads to 1a-Ni, which triggers a 5-exo-trig carbonickelation on the terminal olefin; (ii) the resulting 2a-Ni does not undergo the expected Ni–H elimination but probably evolves by disproportionation [20] leading to alkyl2Ni and NiBr2bipy [21,22]. The subsequent reductive elimination of alkyl2Ni would explain the formation of the dimer 4a.
Table 1: Carbonickelation of compound 1a–c.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-81-i7.svg?max-width=637&scale=1.0)
|
|||
| entry | ArI | Z | ratio 2/2’/3/4a |
|---|---|---|---|
| 1 | 1a | O | ε/ε/ε/100 (26%b) |
| 2 | 1b | N-Boc | 0/50/50/0 (60%) |
| 3 | 1c | N-Ms | 61/26/13/0 (60%c) |
| 4d | 1c | N-Ms | 40/22/38/0 (69%c) |
aDetermined by NMR, isolated yield between brackets; btrace amounts of 2a, 2a’ and 3a were identified as well as several other byproducts; cNMR yield with respect to an internal standard, based on initial aryl iodide 1c; dreaction run with 1 equiv of NiBr2bipy.
In contrast, the carbonickelation of 1b led to an equimolar mixture of the expected 3-methylindole 2b’ (3-methyleneindoline 2b rearranging into 3-methylindol 2b’, probably during work-up) and 3-methylindoline 3b in an overall good 60% isolated yield (Table 1, entry 2). The formation of 3 is probably due to the sluggishness of the NiH elimination, which allows for the competitive protonation of the fragile intermediate alkylnickel 2-Ni. While the Pd-catalyzed reductive Heck reaction promoted by a hydride generated in situ is well known [23,24], the nickel-catalyzed process is likely to occur through a radical hydrogen transfer from the DMF [20,25]. The N-allylaniline 1c gives the same good yield added to an attractive (2c + 2c’)/3c ratio of 87/13 (Table 1, entry 3). Formation of the 3-methylindoline 3 is therefore disfavored when a mesyl protecting group is used instead of a carbamate. When this reaction is run with stoichiometric amounts of nickel, the reductive pathway affording indoline 3c is slightly increased, and a (2c + 2c’)/3c ratio of 62/38 is observed (Table 1, entry 4).
In conclusion to the first part of this study, the formation and cyclization of arylnickel intermediates 1-Ni is observed in all cases. Afterward, the stability of the exo-methylene-nickel 2-Ni seems to govern the formation of the cyclized product. Particularly, the dimerization of 2-Ni threatens the synthetic utility of this reaction, as observed in the case of 1a. In an effort to escape this pathway, we tried to stabilize 2-Ni by using allyl moieties that would provide secondary alkylnickel intermediates. Crotyl and cyclohexenyl ethers and amines were thus employed instead of the allyl. Compounds 5 and 6 were easily prepared by a Mitsunobu condensation involving 2-iodophenol or 2-iodo-N-mesylaniline and crotyl alcohol or cyclohex-2-enol (Scheme 2). An N-mesyl derivative was retained, with a lesser amount of indoline 3 being obtained above when this protecting group was employed.
![[1860-5397-9-81-i2]](/bjoc/content/inline/1860-5397-9-81-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of substrates 5a, 5c, 6a and 6c.
Scheme 2: Synthesis of substrates 5a, 5c, 6a and 6c.
The carbonickelation protocol was applied to the cyclization of crotyl derivatives 5 (Scheme 3).
![[1860-5397-9-81-i3]](/bjoc/content/inline/1860-5397-9-81-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cyclization of substrate 5a and 5c.
Scheme 3: Cyclization of substrate 5a and 5c.
When applied to ether 5a, as expected, the substitution of the allyl moiety at the terminal position by a methyl tends to disfavor the dimerization of the alkylnickel intermediate, and type-4 dimers are no longer observed. However, the expected cyclized compound 7a is obtained only in trace amounts, with the major products isolated being compounds 8a and 9a. Formation of these unexpected products could result from the consecutive intermolecular reactions between 5a and the alkylnickel 7a-Ni or the arylnickel 5a-Ni, respectively (Scheme 4). This reactivity is not unexpected: allyl ethers are known to be good allylating agents in the presence of nickel-bipyridine complexes [26,27]. In addition, the electroreductive allylation of aromatic or heteroaromatic halide and allylic acetate [28,29] was successfully carried out using nickel-bipyridine complexes as catalysts in DMF. More recently, Weix noted that the same reaction could be achieved under chemical conditions, always using bi-(or ter-)pyridine nickel catalysts [30]. These data explain the formation of 9a. Finally, the 8a/9a ratio suggests that 5a-Ni can undergo two competitive pathways: the 5-exo-trig cyclization leading to 7a-Ni, and the aromatic allylation affording 9a. Based on the figures we conclude that 5a-Ni reacts more rapidly with the allyl derivative than it cyclizes.
![[1860-5397-9-81-i4]](/bjoc/content/inline/1860-5397-9-81-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism involving π-allylnickel formation.
Scheme 4: Proposed mechanism involving π-allylnickel formation.
Under the same conditions, N-mesylaniline 5c is more efficient (Scheme 3), and the expected cyclized product 7c is obtained as the major product (60% GC yield, 31% isolated yield due to the instability of the exocyclic double bond during the purification) [31].
In the cyclohexenyl series, we were pleased to observe that ether 6a affords only the benzofuran 10a, but the conversion is limited (30% of the starting material 6a is recovered) and the yield modest (36% isolated, 51% based on recovered material, Scheme 5).
![[1860-5397-9-81-i5]](/bjoc/content/inline/1860-5397-9-81-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Cyclization of substrate 6a and 6c.
Scheme 5: Cyclization of substrate 6a and 6c.
Interestingly, the intracyclic character of the double bond in 6 appears to influence the relative kinetics of the competing reactions in favor of both the carbonickelation and the β-elimination (thus the Heck-type coupling) over the formation of a π-allyl complex that would afford allylation products such as 8 or 9. In the aniline series, the indole 10c (50% GC yield, 28% isolated yield) is obtained together with the indoline 11c, in an indol/indoline 10c/11c ratio of 80/20.
Creation of an all-carbon quaternary center at a ring junction
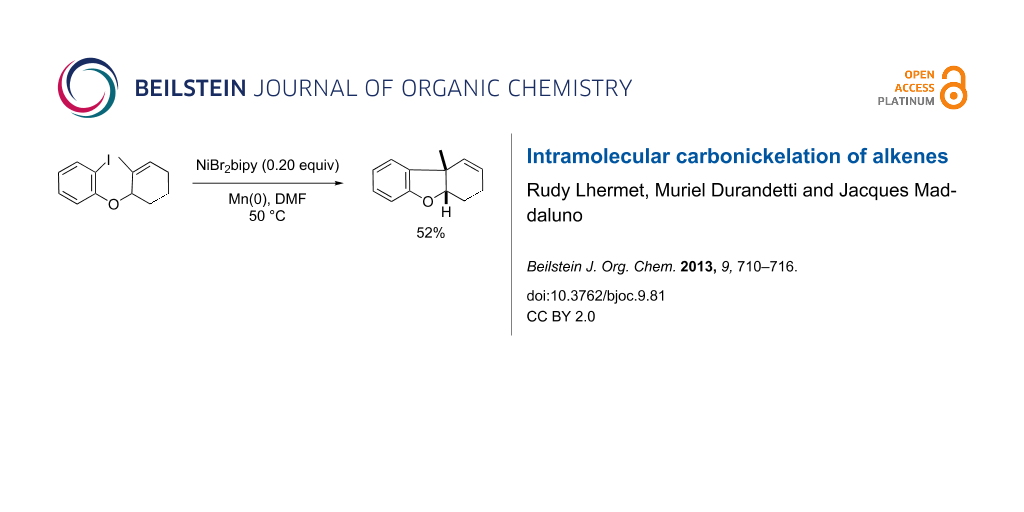
The success of the above experiments prompted us to conduct this reaction with trisubstituted olefins, in an effort to construct tricyclic skeletons with an all-carbon quaternary center at a ring junction. This pattern is found in important natural products such as morphine whose ACE ring system exhibits a tetrahydrodibenzofuran motif with an angular ethylamino chain on C13. We thought that the nickel-catalyzed intramolecular carbometalation reaction could help to tackle the problem of the central ring (E) closure. To validate this hypothesis, we retained substrate 13 as a simplified working model. If ether 13 is little functionalized, it bears the methylvinyl moiety that is essential to the construction of the quaternary ring junction. Substrate 13 was prepared by a simple Mitsunobu condensation between 2-methylcyclohex-2-enol (12), a known compound prepared in three steps from commercially available 2-methylcyclohexanone and 2-iodophenol (Scheme 6). The carbonickelation protocol applied to 13 led in one hour to the expected (and sole) tricyclic product 14 in 52% isolated yield, resulting from a nickel-catalyzed intramolecular base-free Heck-type coupling and exhibiting an all-carbon quaternary center at a cis-ring junction (as established by NOESY experiments). This result, added to that obtained above with aryl ether 6a, underlines that the endocyclic character of the unsaturation is essential to favor the Heck coupling. The carbonickelation process leads to a secondary alkylnickel intermediate, which is sufficiently stabilized to avoid the side reactions observed above. Actually, the carbonickelation remains the rate-determining step as suggested by an experiment in which allyl acetate was mixed with 13 before the addition of the catalyst (Barbier conditions). In this case, the only product was the allylated aryl derivative 17, recovered in comparable yields, suggesting that in the presence of a good allyl donor, the σ-arylnickel undergoes an intermolecular allylation quicker than the intramolecular carbonickelation.
![[1860-5397-9-81-i6]](/bjoc/content/inline/1860-5397-9-81-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis and carbometalations of 13.
Scheme 6: Synthesis and carbometalations of 13.
From a synthetic point of view, it is interesting to note that 14 was obtained as only one isomer, the double bond remaining at the location imposed by the nickel hydride elimination. In order to compare the Ni-catalyzed version to the Pd(0) one, the same reaction was run following the protocol recently described by Fukuyama [32] in the key-step of his total synthesis of morphine. Under these conditions, the cyclization took place in an excellent 82% yield after 3.5 h, but a mixture of the three regioisomers 14–16 was recovered (Scheme 6). By contrast, under Larock’s conditions [33] [Pd(OAc)2 (5 mol %), Na2CO3, DMF, 80 °C], an even higher yield (90% after 2 days) was returned but consisted of a 1:1:1 mixture of the same three products 14–16. Replacing sodium carbonate by silver carbonate avoids these post-cyclization isomerizations [34,35], but the yield (52% after 16 h) was not higher than with nickel. Thus, this study suggests that a Heck-coupling reaction relying on a carbonickelation step can be considered as a useful tool in the total synthesis.
Conclusion
In this work, we have shown that the NiBr2bipy complex can be used to catalyze an intramolecular Heck-type reaction in the absence of any additional base. This glove-box-free procedure occurs using 20% of NiBr2bipy and does not require the handling of air- or moisture-sensitive reagents. Thus, this single process gives access to a simplified model of the trinuclear ACE core of morphine. Beyond the appeal of the possible replacement of expensive palladium by cheap nickel, the absence of post-coupling isomerization of the double bond seems particularly worthy of note.
Experimental
General procedure for the intramolecular carbonickelation of alkenes
To a solution of aryliodide (0.5–1 mmol, 1 equiv) in anhydrous DMF (5 mL) under argon atmosphere at 50 °C is added manganese (2 equiv) followed by NiBr2bipy (0.2 equiv) then rapidly TFA (20 μL). The medium is vigorously stirred at 50 °C, and disappearance of the starting material is monitored by gas chromatography. The mixture is hydrolyzed with water (10 mL), diluted with Et2O (10 mL), and then filtered through celite. The aqueous layer is extracted with Et2O (2 × 10 mL), and then the combined organic layers are washed with water (3 × 10 mL) and brine (2 × 10 mL), dried over anhydrous MgSO4, and concentrated. The crude is purified by flash chromatography.
cis-9b-methyl-3,4,4a,9b-tetrahydrodibenzo[b,d]furan (14)
The compound 14 is obtained by using ether 13 (314 mg, 1 mmol), NiBr2bipy (75 mg, 0.2 mmol), and manganese powder (110 mg, 2 mmol) in anhydrous DMF (5 mL) following the carbonickelation procedure. The pure 14 (97 mg, 0.52 mmol, 52%) is isolated from the crude by flash chromatography on silica (2% of Et2O in n-pentane) as a colorless oil. 1H NMR (300 MHz, CDCl3) 1.41 (s, 3H), 1.78–2.03 (m, 2H), 2.18–2.31 (m, 2H), 4.62 (t, J = 3.6 Hz, 1H), 5.51–5.56 (m, 1H), 5.68–5.75 (m, 1H), 6.79 (dd, J = 8.4, 1.0 Hz, 1H), 6.87 (td, J = 7.2, 0.9 Hz, 1H), 7.11 (d, J = 7.2 Hz, 1H), 7.12 (td, J = 6.6, 1.2 Hz, 1H); 13C NMR (75 MHz, CDCl3) 19.4, 23.3, 25.1, 44.4, 87.8, 110.0, 120.7, 122.9, 125.5, 128.1, 132.1, 135.8, 158.9; NMR 2D NOESY: correlation between 1.41 (s, 3H) and 4.62 (t, J = 3.6 Hz, 1H); IR (neat): 3018, 2956, 1595, 1474, 1232, 1039 cm−1; MS (CI) m/z: 186 (M+), 171 (M − Me, base), 143, 128; HRMS (EI): calcd for (M+) C13H14O: 186.1045; found: 186.1049.
Supporting Information
| Supporting Information File 1: Experimental procedures and compound characterization. | ||
| Format: PDF | Size: 254.3 KB | Download |
References
-
Marek, I.; Chinkov, N.; Banon-Tenne, D. Carbometallation Reactions. In Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; de Meijere, A.; Diederich, F., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004; pp 395–478. doi:10.1002/9783527619535.ch7
Return to citation in text: [1] -
Hogan, A.-M. L.; O’Shea, D. F. Chem. Commun. 2008, 3839–3851. doi:10.1039/b805595e
Return to citation in text: [1] -
Ziegler, K.; Bähr, K. Chem. Ber. 1928, 61, 253–263. doi:10.1002/cber.19280610203
Return to citation in text: [1] -
Heck, R. F.; Nolley, J. P. J. Org. Chem. 1972, 37, 2320–2322. doi:10.1021/jo00979a024
Return to citation in text: [1] -
Heck, R. F. Acc. Chem. Res. 1979, 12, 146–151. doi:10.1021/ar50136a006
Return to citation in text: [1] -
Beletskaya, I. P.; Cheprakov, A. V. Chem. Rev. 2000, 100, 3009–3066. doi:10.1021/cr9903048
Return to citation in text: [1] -
Dounay, A. B.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2964. doi:10.1021/cr020039h
See for a review on palladium-catalyzed reactions involving the Mizoroki–Heck reaction.
Return to citation in text: [1] -
Negishi, E.-i.; Copéret, C.; Ma, S.; Liou, S.-Y.; Liu, F. Chem. Rev. 1996, 96, 365–394. doi:10.1021/cr950020x
Return to citation in text: [1] -
Ma, S.; Negishi, E.-i. J. Org. Chem. 1994, 59, 4730–4732. doi:10.1021/jo00096a011
Return to citation in text: [1] -
Taniguchi, T.; Zaimoku, H.; Ishibashi, H. J. Org. Chem. 2009, 74, 2624–2626. doi:10.1021/jo802787j
Return to citation in text: [1] -
Oestreich, M. The Mizoroki-Heck reaction; John Wiley & Sons, Ltd: Chichester, U.K., 2009.
Return to citation in text: [1] -
Rollin, Y.; Meyer, G.; Troupel, M.; Fauvarque, J.-F.; Périchon, J. J. Chem. Soc., Chem. Commun. 1983, 793–794. doi:10.1039/C39830000793
Return to citation in text: [1] -
Olivero, S.; Rolland, J.-P.; Duñach, E. Organometallics 1998, 17, 3747–3753. doi:10.1021/om980247t
Return to citation in text: [1] -
Ma, S.; Wang, H.; Gao, K.; Zhao, F. J. Mol. Catal. A: Chem. 2006, 248, 17–20. doi:10.1016/j.molcata.2005.12.013
Return to citation in text: [1] -
Gøgsig, T. M.; Kleimark, J.; Lill, S. O. N.; Korsager, S.; Lindhardt, A. T.; Norrby, P.-O.; Skrydstrup, T. J. Am. Chem. Soc. 2012, 134, 443–452. doi:10.1021/ja2084509
Return to citation in text: [1] -
Durandetti, M.; Gosmini, C.; Périchon, J. Tetrahedron 2007, 63, 1146–1153. doi:10.1016/j.tet.2006.11.055
Return to citation in text: [1] -
Gosmini, C.; Bassene-Ernst, C.; Durandetti, M. Tetrahedron 2009, 65, 6141–6146. doi:10.1016/j.tet.2009.05.044
Return to citation in text: [1] -
Durandetti, M.; Hardou, L.; Lhermet, R.; Rouen, M.; Maddaluno, J. Chem.–Eur. J. 2011, 17, 12773–12783. doi:10.1002/chem.201100967
Return to citation in text: [1] -
Durandetti, M.; Hardou, L.; Clément, M.; Maddaluno, J. Chem. Commun. 2009, 4753–4755. doi:10.1039/b902095k
Return to citation in text: [1] -
Jahn, U. Radicals in Synthesis III. Heinrich, M. R.; Gansäuer, A., Eds.; Topics in Current Chemistry, Vol. 320; Spinger-Verlag: Berlin Heidelberg, Germany, 2012; pp 323–352. doi:10.1007/978-3-642-28123-5
Radical intermediates have been proposed in case of alkylnickel.
Return to citation in text: [1] [2] -
De França, K. W. R.; Navarro, M.; Léonel, E.; Durandetti, M.; Nédélec, J.-Y. J. Org. Chem. 2002, 67, 1838–1842. doi:10.1021/jo016280y
Return to citation in text: [1] -
Jutand, A. Chem. Rev. 2008, 108, 2300–2347. doi:10.1021/cr068072h
Return to citation in text: [1] -
Burns, B.; Grigg, R.; Sridharan, V.; Worakun, T. Tetrahedron Lett. 1988, 29, 4325–4328. doi:10.1016/S0040-4039(00)80488-8
Return to citation in text: [1] -
Gao, P.; Cook, S. P. Org. Lett. 2012, 14, 3340–3343. doi:10.1021/ol3013167
See for a recent application.
Return to citation in text: [1] -
Tsou, T. T.; Kochi, J. J. Am. Chem. Soc. 1979, 101, 6319–6332. doi:10.1021/ja00515a028
Return to citation in text: [1] -
Olivero, S.; Duñach, E. Synlett 1994, 531–533. doi:10.1055/s-1994-22917
Return to citation in text: [1] -
Franco, D.; Wenger, K.; Antonczak, S.; Cabrol-Bass, D.; Duñach, E.; Rocamora, M.; Gomez, M.; Muller, G. Chem.–Eur. J. 2002, 8, 664–672. doi:10.1002/1521-3765(20020201)8:3<664::AID-CHEM664>3.0.CO;2-D
Return to citation in text: [1] -
Durandetti, M.; Nédélec, J.-Y.; Périchon, J. J. Org. Chem. 1996, 61, 1748–1755. doi:10.1021/jo9518314
Return to citation in text: [1] -
Durandetti, M.; Périchon, J. Synthesis 2004, 3079–3083. doi:10.1055/s-2004-834896
Return to citation in text: [1] -
Anka-Lufford, L. L.; Prinsell, M. R.; Weix, D. J. J. Org. Chem. 2012, 77, 9989–10000. doi:10.1021/jo302086g
Return to citation in text: [1] -
Attempts to minimize the plummeting of the yield upon purification using other chromatographic conditions (in particular replacing silica by various alumina) proved unsuccessful on comparable 3-methylidenetetrahydrobenzofurans.
Return to citation in text: [1] -
Uchida, K.; Yokoshima, S.; Kan, T.; Fukuyama, T. Org. Lett. 2006, 8, 5311–5313. doi:10.1021/ol062112m
Return to citation in text: [1] -
Larock, R. C.; Stinn, D. E. Tetrahedron Lett. 1988, 29, 4687–4690. doi:10.1016/S0040-4039(00)80581-X
Return to citation in text: [1] -
Parsons, P. J.; Charles, M. D.; Harvey, D. M.; Sumoreeah, L. R.; Shell, A.; Spoors, G.; Gill, A. L.; Smith, S. Tetrahedron Lett. 2001, 42, 2209–2211. doi:10.1016/S0040-4039(01)00111-3
Return to citation in text: [1] -
Trost, B. M.; Tang, W.; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 14785–14803. doi:10.1021/ja054449+
Return to citation in text: [1]
| 1. | Marek, I.; Chinkov, N.; Banon-Tenne, D. Carbometallation Reactions. In Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; de Meijere, A.; Diederich, F., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004; pp 395–478. doi:10.1002/9783527619535.ch7 |
| 2. | Hogan, A.-M. L.; O’Shea, D. F. Chem. Commun. 2008, 3839–3851. doi:10.1039/b805595e |
| 6. | Beletskaya, I. P.; Cheprakov, A. V. Chem. Rev. 2000, 100, 3009–3066. doi:10.1021/cr9903048 |
| 20. |
Jahn, U. Radicals in Synthesis III. Heinrich, M. R.; Gansäuer, A., Eds.; Topics in Current Chemistry, Vol. 320; Spinger-Verlag: Berlin Heidelberg, Germany, 2012; pp 323–352. doi:10.1007/978-3-642-28123-5
Radical intermediates have been proposed in case of alkylnickel. |
| 21. | De França, K. W. R.; Navarro, M.; Léonel, E.; Durandetti, M.; Nédélec, J.-Y. J. Org. Chem. 2002, 67, 1838–1842. doi:10.1021/jo016280y |
| 22. | Jutand, A. Chem. Rev. 2008, 108, 2300–2347. doi:10.1021/cr068072h |
| 4. | Heck, R. F.; Nolley, J. P. J. Org. Chem. 1972, 37, 2320–2322. doi:10.1021/jo00979a024 |
| 18. | Durandetti, M.; Hardou, L.; Lhermet, R.; Rouen, M.; Maddaluno, J. Chem.–Eur. J. 2011, 17, 12773–12783. doi:10.1002/chem.201100967 |
| 3. | Ziegler, K.; Bähr, K. Chem. Ber. 1928, 61, 253–263. doi:10.1002/cber.19280610203 |
| 19. | Durandetti, M.; Hardou, L.; Clément, M.; Maddaluno, J. Chem. Commun. 2009, 4753–4755. doi:10.1039/b902095k |
| 12. | Rollin, Y.; Meyer, G.; Troupel, M.; Fauvarque, J.-F.; Périchon, J. J. Chem. Soc., Chem. Commun. 1983, 793–794. doi:10.1039/C39830000793 |
| 13. | Olivero, S.; Rolland, J.-P.; Duñach, E. Organometallics 1998, 17, 3747–3753. doi:10.1021/om980247t |
| 16. | Durandetti, M.; Gosmini, C.; Périchon, J. Tetrahedron 2007, 63, 1146–1153. doi:10.1016/j.tet.2006.11.055 |
| 11. | Oestreich, M. The Mizoroki-Heck reaction; John Wiley & Sons, Ltd: Chichester, U.K., 2009. |
| 17. | Gosmini, C.; Bassene-Ernst, C.; Durandetti, M. Tetrahedron 2009, 65, 6141–6146. doi:10.1016/j.tet.2009.05.044 |
| 8. | Negishi, E.-i.; Copéret, C.; Ma, S.; Liou, S.-Y.; Liu, F. Chem. Rev. 1996, 96, 365–394. doi:10.1021/cr950020x |
| 9. | Ma, S.; Negishi, E.-i. J. Org. Chem. 1994, 59, 4730–4732. doi:10.1021/jo00096a011 |
| 10. | Taniguchi, T.; Zaimoku, H.; Ishibashi, H. J. Org. Chem. 2009, 74, 2624–2626. doi:10.1021/jo802787j |
| 7. |
Dounay, A. B.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2964. doi:10.1021/cr020039h
See for a review on palladium-catalyzed reactions involving the Mizoroki–Heck reaction. |
| 14. | Ma, S.; Wang, H.; Gao, K.; Zhao, F. J. Mol. Catal. A: Chem. 2006, 248, 17–20. doi:10.1016/j.molcata.2005.12.013 |
| 15. | Gøgsig, T. M.; Kleimark, J.; Lill, S. O. N.; Korsager, S.; Lindhardt, A. T.; Norrby, P.-O.; Skrydstrup, T. J. Am. Chem. Soc. 2012, 134, 443–452. doi:10.1021/ja2084509 |
| 26. | Olivero, S.; Duñach, E. Synlett 1994, 531–533. doi:10.1055/s-1994-22917 |
| 27. | Franco, D.; Wenger, K.; Antonczak, S.; Cabrol-Bass, D.; Duñach, E.; Rocamora, M.; Gomez, M.; Muller, G. Chem.–Eur. J. 2002, 8, 664–672. doi:10.1002/1521-3765(20020201)8:3<664::AID-CHEM664>3.0.CO;2-D |
| 23. | Burns, B.; Grigg, R.; Sridharan, V.; Worakun, T. Tetrahedron Lett. 1988, 29, 4325–4328. doi:10.1016/S0040-4039(00)80488-8 |
| 24. |
Gao, P.; Cook, S. P. Org. Lett. 2012, 14, 3340–3343. doi:10.1021/ol3013167
See for a recent application. |
| 20. |
Jahn, U. Radicals in Synthesis III. Heinrich, M. R.; Gansäuer, A., Eds.; Topics in Current Chemistry, Vol. 320; Spinger-Verlag: Berlin Heidelberg, Germany, 2012; pp 323–352. doi:10.1007/978-3-642-28123-5
Radical intermediates have been proposed in case of alkylnickel. |
| 25. | Tsou, T. T.; Kochi, J. J. Am. Chem. Soc. 1979, 101, 6319–6332. doi:10.1021/ja00515a028 |
| 33. | Larock, R. C.; Stinn, D. E. Tetrahedron Lett. 1988, 29, 4687–4690. doi:10.1016/S0040-4039(00)80581-X |
| 34. | Parsons, P. J.; Charles, M. D.; Harvey, D. M.; Sumoreeah, L. R.; Shell, A.; Spoors, G.; Gill, A. L.; Smith, S. Tetrahedron Lett. 2001, 42, 2209–2211. doi:10.1016/S0040-4039(01)00111-3 |
| 35. | Trost, B. M.; Tang, W.; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 14785–14803. doi:10.1021/ja054449+ |
| 31. | Attempts to minimize the plummeting of the yield upon purification using other chromatographic conditions (in particular replacing silica by various alumina) proved unsuccessful on comparable 3-methylidenetetrahydrobenzofurans. |
| 32. | Uchida, K.; Yokoshima, S.; Kan, T.; Fukuyama, T. Org. Lett. 2006, 8, 5311–5313. doi:10.1021/ol062112m |
| 28. | Durandetti, M.; Nédélec, J.-Y.; Périchon, J. J. Org. Chem. 1996, 61, 1748–1755. doi:10.1021/jo9518314 |
| 29. | Durandetti, M.; Périchon, J. Synthesis 2004, 3079–3083. doi:10.1055/s-2004-834896 |
| 30. | Anka-Lufford, L. L.; Prinsell, M. R.; Weix, D. J. J. Org. Chem. 2012, 77, 9989–10000. doi:10.1021/jo302086g |
© 2013 Lhermet et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)