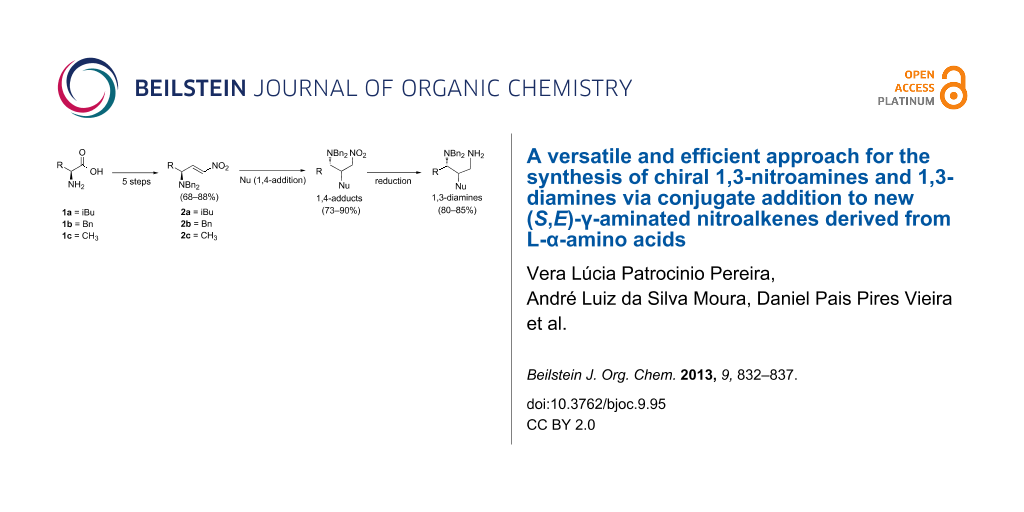
Abstract
New chiral (S,E)-γ-N,N-dibenzylated nitroalkenes 2a–c were synthesized from natural L-(α)-amino acids in five steps with overall yields of 68–88%. The conjugate addition of hydride, methoxide, nitronate and azide nucleophiles to 2a–c led to the corresponding chiral 1,3-nitroamines in 74–90% yield. The conjugate addition of cyanide anion to 2a,b was followed by HNO2 elimination affording chiral aminated acrylonitriles (73–98%). On the other hand, the azide anion reacted with 2a, in acetonitrile, via a [3 + 2]-cycloaddition in which HNO2 was lost, providing the corresponding 1,2,3-triazole derivative. Direct reduction of 1,3-nitroamine derivatives 9a,b produced the corresponding 1,3-diamines in good yields.

Graphical Abstract
Introduction
Nitroalkenes constitute a class of organic compounds that present exceptional versatility in organic synthesis [1-4]. They are reactive in Michael reactions with a wide variety of nucleophiles [5-10], in Friedel–Crafts alkylations [11-14], and Baylis–Hillman reactions [15-18]. Furthermore, nitroalkenes can react as dipolarophiles in [3 + 2] cycloadditions [19-22], as dienophiles in [4 + 2] cycloadditions [23-26], and as heterodienes in hetero-Diels–Alder reactions [27-31] and even participate in cross-coupling reactions [32]. Due to this notable reactivity, obtaining new chiral and achiral nitroalkenes is of great importance in synthetic organic chemistry. Achiral nitroalkenes, specifically the chemically stable β-trans-nitrostyrene and its phenyl-substituted analogues, besides the heteroaromatic nitroalkenes, have been employed widely in highly enantioselective syntheses promoted by chiral metallic catalysts [33-37] and most recently by a great number of different chiral organocatalysts [38-41] or combinations of both [42-44]. Nonetheless, the use of chiral nitroalkenes bearing a stereogenic center in the γ-position in nonracemic diastereoselective syntheses ("chiron" approach [45]) is less frequent in the literature. Notably, chiral nitroalkenes derived from carbohydrates [46-50] have been most often utilized as "chirons" in diastereoselective syntheses. Most nitroalkenes are synthesized from a nitroaldol product followed by elimination of the activated hydroxy group in the sulfonate or acetate form [51] although various routes have often been applied [52-54]. On the other hand, chiral nitroamines [55,56] are important intermediates directly transformed, via reduction of the nitro group, into diamines [57] or via a Nef reaction in amino acids [58]. Specifically, chiral 1,2-nitroamines have been mainly accessed by direct nucleophilic addition of nitronate anions to imine derivatives by the use of chiral metal and organocatalysts [55,56]. In contrast, chiral 1,3-nitroamines, despite constituting direct precursors of chiral 1,3-diamines, are scarcely synthesized by conjugate addition of nitroalkanes to nitroalkenes. Chiral 1,3-diamines present extraordinary opportunities in enantioselective synthesis as chiral auxiliaries, chiral catalysts, chiral reagents and chiral ligands [59,60]. In our continuing interest in synthesizing new chiral nonracemic nitro compounds [61-68] by a "chiron" approach [45], we intended to develop a route for obtaining γ-nitrogenated chiral nonracemic nitroalkenes 2a–c from L-leucine (1a), L-phenylalanine (1b) and L-alanine (1c), respectively, and investigate their reactivities in conjugate additions with different nucleophiles as a way of yielding chiral nonracemic 1,3-nitroamine and its corresponding 1,3-diamine derivatives.
Results and discussion
The preparation of 2a–c (Scheme 1) was initiated with the synthesis of Reetz’s α-aminoaldehydes [69,70]. In this manner, the α-amino acids 1a–c were perbenzylated using a slight modification in the experimental procedures described by Beaulieu [71] and Warren [72] leading to tribenzylated esters 3a–c in 90–99% isolated yield. Next, reduction of 3a–c with LiAlH4 provided the desired alcohols 4a–c (87–95% yield), which were oxidized under Swern conditions to aldehydes 5a–c in 96–99% yield. These were submitted to a Henry reaction, according to the modified conditions developed by Hanessian [73]. Thus, 5a–c were reacted with nitromethane, at room temperature (instead of 0 °C) in the presence of 0.2 equivalents of TBAF·3H2O (instead of 1 equivalent) as base, furnishing an anti:syn-nitroalcohol mixture 6a–c in 91–95% yield and 68–86% d.e. The activation of the hydroxy groups of 6a–c by transformation into the corresponding mesylates was followed by smooth elimination at −78 °C, leading to the desired nitroalkenes 2a–c in 95–99% yield, as single E-diastereoisomers. It is worth mentioning that the nitroalkenes 2a–c are obtained in high chemical and stereochemical purity and can be stored in a freezer or even at room temperature for several months without decomposition occurring.
![[1860-5397-9-95-i1]](/bjoc/content/inline/1860-5397-9-95-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: (i) K2CO3, EtOH/H2O 5:1, BnBr, 100 °C, 12 h (90–99%). (ii) (a) LiAlH4, THF dry, 100 °C, 8 h. (b) NaOHaq (87–95%). (iii) (a) (COCl)2, CH2Cl2, DMSO, −78 °C, 30 min, then TEA, −78 °C to rt (96–99%). (iv) TBAF·3H2O (0.2 equiv), CH3NO2, THFdry, rt, 4 h (91–95%). (v) (a) MsCl, CH2Cl2, −78 °C, then TEA 60 min, rt (95–99 %).
Scheme 1: Reagents and conditions: (i) K2CO3, EtOH/H2O 5:1, BnBr, 100 °C, 12 h (90–99%). (ii) (a) LiAlH4, THF...
In order to verify the enantiomeric purity of the new nitroalkenes 2a–c and to confirm their stereochemical stability, a chiral HPLC analysis was conducted. Racemic (+/−)-2b was synthesized from (D/L)-phenylalanine by the same route employed for (−)-2b and utilized as standard (Figure 1). The racemic nitroalkene (+/−)-2b showed a very good separation factor in the chromatograph chiral column used. Figure 1 shows that (−)-2b prepared from L-phenylalanine was enantiomerically pure (enantiomeric excess > 99%).
![[1860-5397-9-95-1]](/bjoc/content/figures/1860-5397-9-95-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chiral HPLC analysis: Chiralpak AD-H; n-hexane/2-propanol 99:1 (0.6 mL/min), T = 25 °C; UV–vis detection at λ = 254 nm; retention times: (+/−)-2 (11.13 and 11.60 min) and (−)-2b (10.99 min).
Figure 1: Chiral HPLC analysis: Chiralpak AD-H; n-hexane/2-propanol 99:1 (0.6 mL/min), T = 25 °C; UV–vis dete...
HPLC analysis showed that the stereochemical integrity of 2 was preserved throughout the synthetic process. Next, the reactivity of the new chiral nitroalkenes was investigated with respect to the conjugate addition of hydride, nitronate, methoxide, cyanide, and azide anions in addition to benzylamine aiming to obtain directly 1,3-nitroamine derivatives. All the experiments were carried out at room temperature and the results are summarized in Table 1.
Table 1: Reactivity of 2a–c with different nucleophiles.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-95-i3.svg?max-width=637&scale=1.0)
|
||||||
| entry | nitroalkene | Nu | base/solvent/time (h) | adduct | yield (%) | anti/syna |
|---|---|---|---|---|---|---|
| 1 | 2b | NaBH4 | –/CHCl3–iPrOH (16:3)/1 | 7b | 85 | – |
| 2 | 2a | CH3NO2 | DBU 0.3 equiv/CH3CN/12 | 8a | 86 | – |
| 3 | 2b | CH3NO2 | TBAF·3H2O/THF/8 | 8b | 80 | – |
| 4 | 2a | LiOMeb | –/MeOH/0.5 | 9a | 90 | 94:6 |
| 5 | 2b | LiOMeb | –/THF/0.5 | 9b | 85 | 94:6 |
| 6 | 2a | LiOMeb | –/MeOH/24 | 9a | 90 | 50:50 |
| 7 | 2b | LiOMeb | –/MeOH/20 | 9b | 70 | 57:43 |
| 8 | 2c | LiOMeb | –/MeOH/12 | 9c | 77 | 69:31 |
| 9 | 2a | TMSCN | TBAF·3H2O/CH3CN/6 | 12a | 98 | – |
| 10 | 2b | TMSCN | TBAF·3H2O/CH3CN/12 | 12b | 73 | – |
| 11 | 2b | TMSN3 | TBAF·3H2O/THF/16 | 10b | 76 | 86:14 |
| 12 | 2c | TMSN3 | TBAF·3H2O/THF/20 | 10c | 73 | 74:26 |
| 13 | 2a | TMSN3 | TBAF·3H2O/CH3CN/16 | 11a | 74 | – |
| 14 | 2a | BnNH2 excess | –/–/48 | –c | – | – |
| 15 | 2b | BnNH2 | DBU 0.5 equiv/CH3CN/8 | –c | – | – |
aDiastereoisomeric ratio. bSolution 1 M in MeOH. cNo reaction observed.
The selective conjugate addition of hydride anion to 2b produced the chiral 1,3-nitroamine 7b in 85% yield [74] (Table 1, entry 1). Similarly, the addition of nitromethane to 2a,b led to the corresponding 2-nitromethyl-1,3-nitroamine derivatives 8a,b in good yields (Table 1, entries 2 and 3). Other nitronate anions also reacted adequately. On the other hand, a literature search showed that the addition of an oxygenated nucleophile to nitroalkenes has been scarcely studied due to the low reactivity of neutral nucleophiles associated with the reaction reversibility [46,75,76]. Addition of methoxide anion to 2a–c led to the corresponding chiral 2-methoxy-1,3-nitroamine derivatives 9a–c in 70–90% yield (Table 1, entries 4–8). A high d.r. was obtained when shorter reaction times were employed (Table 1, entries 4 and 5). On the other hand, a severe decrease in the diastereoselectivity occurred with longer durations, presumably due to an equilibration by a retro-conjugate addition [77] (Table 1, entries 6–8). Next, the conjugate cyanide addition to 2a,b promoted by TMSCN 1 equiv/TBAF·3H2O 1 equiv led to 12a,b in 98 and 73% respectively, instead of 2-cyano-1,3-nitroamines derivatives (Table 1, entries 9 and 10). Products 12a,b were likely formed from the conjugate addition of cyanide to 2a,b followed by HNO2 elimination. It is worthy of note that the Michael addition of “HCN” equivalent to nitroalkenes is rare in the literature [78-83]. Paulsen [78] and Sakakibara [79] were the first authors to report the cyanide addition to α,β-unsaturated nitroalkenes. In agreement with our results, Sakakibara [79] and Spanevello [80] observed a total or partial HNO2 elimination from vicinal nitronitrile adducts. This behavior was not observed in other findings reported [78,81-83] and the corresponding vicinal nitronitriles were the sole products. It is important to note that 12a,b can in theory also be obtained by an aza-Baylis–Hillman reaction [15-18,68]. Thus, this cyanide Michael addition to 2a,b constitutes a synthetic alternative to the cases where N-protected aza-Baylis–Hillman adducts need to be obtained. Interestingly, the addition of “HN3” promoted by TMSN3 1 equiv/TBAF·3H2O 1 equiv furnished either the 2-azido-1,3-nitroamine-derivatives 10b,c (Table 1, entries 11 and 12) or the triazole derivative 11a (Table 1, entry 13) when THF or CH3CN were used as solvents, respectively. The formation of chiral 1,2,3-triazole derivative 11a was proposed to be by way of a [3 + 2]-cycloaddition with the azide group acting as 1,3-dipolarophile, followed by HNO2 elimination leading to aromatization. There are a few reports in the literature where the azide anion is added to α,β-unsaturated nitroalkenes [8,19-22] forming 1,4-adducts [8] or 1,2,3-triazole derivatives [31-33]. Apparently, the reaction course depends on the temperature and concentration of the azide reagent used. In our case, we observed a dependence on the nature of the solvent. However, an influence of the structure of the nitroalkenes has not been independently investigated. Further experiments must be performed for a better understanding of the reaction course. On the other hand, the neat benzylamine addition, or in the presence of DBU (0.5 equiv) as promoter [84], to 2a,b was unsuccessful (Table 1, entries 14 and 15). This nonreactivity can be explained on the basis of the high tendency toward reversibility presented in conjugate additions of amines to reactive nitroalkenes [15,85,86]. As a way of testing the stereochemical stability of the synthesized nitroalkenes in reaction media, 2b was recovered before the reaction was completed (Table 1, entry 3) and the magnitude of the optical rotation of 2b was measured. No loss in enantiomeric purity of 2b was observed in the reaction medium essayed.
Finally, the chiral 1,3-nitroamines 9a,b were easily transformed into the desired chiral 1,3-diamines 14a,b (80% yield) by treatment with NaBH4, NiCl2·6H2O in MeOH [87] (Scheme 2).
![[1860-5397-9-95-i2]](/bjoc/content/inline/1860-5397-9-95-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 1,3-diamines 14a,b. (i) NaBH4/NiCl2·6H2O/MeOH/3 h/rt.
Scheme 2: Synthesis of 1,3-diamines 14a,b. (i) NaBH4/NiCl2·6H2O/MeOH/3 h/rt.
Conclusion
An efficient, reproducible and versatile approach for the synthesis of chiral nonracemic 2-substituted-1,3-nitroamines was developed. The high chemical and stereochemical stability of the new chiral nitroalkenes 2a–c and the variety of 1,3-nitroamines synthesized provide robustness and generality to the methodology. Since the nitroamines 9a,b were efficiently reduced to the corresponding chiral 2-substituted-1,3-diamines 14a,b, the present route constitutes a versatile access to these important classes of substances in high yields.
Supporting Information
| Supporting Information File 1: General information, experimental procedure and spectroscopic data of 3a–c, 6a–c, 2a–c, 7b, 8a, 8b, 11a, 10b–c, 9a–b, 12a, 14a,b. | ||
| Format: PDF | Size: 605.8 KB | Download |
| Supporting Information File 2: NMR, IR and MS spectra. | ||
| Format: PDF | Size: 1.9 MB | Download |
References
-
Barrett, A. G. M. Chem. Soc. Rev. 1991, 20, 95–127. doi:10.1039/cs9912000095
Return to citation in text: [1] -
Ono, N. The Nitro Group in Organic Synthesis; Wiley-VCH: New York, 2001. doi:10.1002/0471224480
Return to citation in text: [1] -
Ballini, R.; Gabrielli, S.; Palmieri, A. Curr. Org. Chem. 2010, 14, 65–83. doi:10.2174/138527210790226429
Return to citation in text: [1] -
Parry, R.; Nishino, S.; Spain, J. Nat. Prod. Rep. 2011, 28, 152–167. doi:10.1039/c0np00024h
Return to citation in text: [1] -
Wu, J.; Mampreian, D. M.; Hoveyda, A. H. J. Am. Chem. Soc. 2005, 127, 4584–4585. doi:10.1021/ja050800f
Return to citation in text: [1] -
Rasappan, R.; Reiser, O. Eur. J. Org. Chem. 2009, 1305–1308. doi:10.1002/ejoc.200801263
Return to citation in text: [1] -
Muruganantham, R.; Namboothiri, I. J. Org. Chem. 2010, 75, 2197–2205. doi:10.1021/jo902595e
Return to citation in text: [1] -
Robiette, R.; Tran, T.-V.; Cordi, A.; Tinant, B.; Marchand-Brynaert, J. Synthesis 2010, 3138–3142. doi:10.1055/s-0030-1258176
Return to citation in text: [1] [2] [3] -
Zhong, C.; Gautam, L. N. S.; Wang, D.; Akhmedov, N. G.; Petersen, J. L.; Shi, X. Tetrahedron 2011, 67, 4402–4411. doi:10.1016/j.tet.2011.01.085
Return to citation in text: [1] -
Rulev, A. Y. Russ. Chem. Rev. 2011, 80, 197–218.
Return to citation in text: [1] -
Poulsen, T. B.; Jørgensen, K. A. Chem. Rev. 2008, 108, 2903–2915. doi:10.1021/cr078372e
Return to citation in text: [1] -
Trost, B. M.; Müller, C. J. Am. Chem. Soc. 2008, 130, 2438–2439. doi:10.1021/ja711080y
Return to citation in text: [1] -
Yuan, Z.-L.; Lei, Z.-Y.; Shi, M. Tetrahedron: Asymmetry 2008, 19, 1339–1346. doi:10.1016/j.tetasy.2008.04.037
Return to citation in text: [1] -
Carmona, D.; Lamata, M. P.; Sánchez, A.; Viguri, F.; Oro, L. A. Tetrahedron: Asymmetry 2011, 22, 893–906. doi:10.1016/j.tetasy.2011.05.003
Return to citation in text: [1] -
Rastogi, N.; Namboothiri, I. N. N.; Cojocaru, M. Tetrahedron Lett. 2004, 45, 4745–4748. doi:10.1016/j.tetlet.2004.04.069
Return to citation in text: [1] [2] [3] -
Deb, I.; Shanbhag, P.; Mobin, S. M.; Namboothiri, I. N. N. Eur. J. Org. Chem. 2009, 4091–4101. doi:10.1002/ejoc.200900475
Return to citation in text: [1] [2] -
Kuan, H.-H.; Reddy, R. J.; Chen, K. Tetrahedron 2010, 66, 9875–9879. doi:10.1016/j.tet.2010.10.061
Return to citation in text: [1] [2] -
Bakthadoss, M.; Sivakumar, N.; Devaraj, A.; Sharada, D. S. Synthesis 2011, 2136–2146. doi:10.1055/s-0030-1260053
Return to citation in text: [1] [2] -
Amantini, D.; Fringuelli, F.; Piermatti, O.; Pizzo, F.; Zunino, E.; Vaccaro, L. J. Org. Chem. 2005, 70, 6526–6529. doi:10.1021/jo0507845
Return to citation in text: [1] [2] -
Quiclet-Sire, B.; Zard, S. Z. Synthesis 2005, 3319–3326. doi:10.1055/s-2005-918463
Return to citation in text: [1] [2] -
Zou, W.; Bhasin, M.; Vembaiyan, K.; Williams, D. T. Carbohydr. Res. 2009, 344, 1024–1027. doi:10.1016/j.carres.2009.03.007
Return to citation in text: [1] [2] -
Arai, T.; Mishiro, A.; Yokoyama, N.; Suzuki, K.; Sato, H. J. Am. Chem. Soc. 2010, 132, 5338–5339. doi:10.1021/ja100265j
Return to citation in text: [1] [2] -
Kim, W. H.; Lee, J. H.; Danishefsky, S. J. J. Am. Chem. Soc. 2009, 131, 12576–12578. doi:10.1021/ja9058926
Return to citation in text: [1] -
Araújo, N.; Gil, M. V.; Román, E.; Serrano, J. A. Tetrahedron 2010, 66, 2664–2674. doi:10.1016/j.tet.2010.02.027
Return to citation in text: [1] -
Mangione, M. I.; Sarotti, A. M.; Suárez, A. G.; Spanevello, R. A. Carbohydr. Res. 2011, 346, 460–464. doi:10.1016/j.carres.2010.12.006
Return to citation in text: [1] -
Butt, N. A.; Moody, C. J. Org. Lett. 2011, 13, 2224–2227. doi:10.1021/ol200477s
Return to citation in text: [1] -
Denmark, S. E.; Thorarensen, A. Chem. Rev. 1996, 96, 137–165. doi:10.1021/cr940277f
Return to citation in text: [1] -
Fringuelli, F.; Matteucci, M.; Piermatti, O.; Pizzo, F.; Burla, M. C. J. Org. Chem. 2001, 66, 4661–4666. doi:10.1021/jo010182v
Return to citation in text: [1] -
Denmark, S. E.; Baiazitov, R. Y. J. Org. Chem. 2006, 71, 593–605. doi:10.1021/jo052001l
Return to citation in text: [1] -
Denmark, S. E.; Baiazitov, R. Y.; Nguyen, S. T. Tetrahedron 2009, 65, 6535–6548. doi:10.1016/j.tet.2009.05.060
Return to citation in text: [1] -
Korotaev, V. Y.; Sosnovskikh, V. Y.; Barabanov, M. A.; Yasnova, E. S.; Ezhikova, M. A.; Kodess, M. I.; Slepukhin, P. A. Tetrahedron 2010, 66, 1404–1409. doi:10.1016/j.tet.2009.11.094
Return to citation in text: [1] [2] -
Denmark, S. E.; Schnute, M. E. J. Org. Chem. 1995, 60, 1013–1019. doi:10.1021/jo00109a037
Return to citation in text: [1] [2] -
Beller, M.; Bolm, C. Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, 2nd ed.; Wiley-VCH: Weinheim, 2004. doi:10.1002/9783527619405
Return to citation in text: [1] [2] -
Jia, Y.-X.; Zhu, S.-F.; Yang, Y.; Zhou, Q.-L. J. Org. Chem. 2006, 71, 75–80. doi:10.1021/jo0516537
Return to citation in text: [1] -
Arai, T.; Yokoyama, N. Angew. Chem., Int. Ed. 2008, 47, 4989–4992. doi:10.1002/anie.200801373
Return to citation in text: [1] -
Gutnov, A. Eur. J. Org. Chem. 2008, 4547–4554. doi:10.1002/ejoc.200800541
Return to citation in text: [1] -
Zhang, W.-H.; Chien, S. W.; Hor, T. S. A. Coord. Chem. Rev. 2011, 255, 1991–2024. doi:10.1016/j.ccr.2011.05.018
Return to citation in text: [1] -
Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r
Return to citation in text: [1] -
List, B. Chem. Rev. 2007, 107, 5413–5415. doi:10.1021/cr078412e
Return to citation in text: [1] -
Bertelsen, S.; Jørgensen, K. A. Chem. Soc. Rev. 2009, 38, 2178–2189. doi:10.1039/b903816g
Return to citation in text: [1] -
Cheong, P. H.-Y.; Legault, C. Y.; Um, J. M.; Çelebi-Ölcüm, N.; Houk, K. N. Chem. Rev. 2011, 111, 5042–5137. doi:10.1021/cr100212h
Return to citation in text: [1] -
Shao, Z.; Zhang, H. Chem. Soc. Rev. 2009, 38, 2745–2755. doi:10.1039/b901258n
Return to citation in text: [1] -
Zhong, C.; Shi, X. Eur. J. Org. Chem. 2010, 2999–3025. doi:10.1002/ejoc.201000004
Return to citation in text: [1] -
Alemán, J.; del Solar, V.; Martín-Santos, C.; Cubo, L.; Ranninger, C. N. J. Org. Chem. 2011, 76, 7287–7293. doi:10.1021/jo2013077
Return to citation in text: [1] -
Hanessian, S.; Franco, J.; Larouche, B. Pure Appl. Chem. 1990, 62, 1887–1910. doi:10.1351/pac199062101887
Return to citation in text: [1] [2] -
Qian, C.; Takayuki, O.; Hasner, A. Synth. Commun. 2000, 30, 293–300. doi:10.1080/00397910008087321
Return to citation in text: [1] [2] -
Hübner, J.; Liebscher, J.; Pätzel, M. Tetrahedron 2002, 58, 10485–10500. doi:10.1016/S0040-4020(02)01406-0
Return to citation in text: [1] -
Sakakibara, T.; Tachmori, Y.; Sudoh, R. Tetrahedron 2004, 40, 1533–1539. doi:10.1016/S0040-4020(01)91801-0
Return to citation in text: [1] -
Fioravanti, S.; Marchetti, F.; Pellacani, L.; Ranieri, L.; Tardella, P. A. Tetrahedron: Asymmetry 2008, 19, 231–236. doi:10.1016/j.tetasy.2007.12.007
Return to citation in text: [1] -
Lehnert, T.; Özüduru, G.; Grugel, H.; Albrecht, F.; Telligmann, S. M.; Boysen, M. M. K. Synthesis 2011, 2685–2708. doi:10.1055/s-0030-1260143
Return to citation in text: [1] -
Melton, J.; McMurry, J. E. J. Org. Chem. 1975, 40, 2138–2139. doi:10.1021/jo00902a033
Return to citation in text: [1] -
Fioravanti, S.; Pellacani, L.; Tardella, P. A.; Vergari, M. C. Org. Lett. 2008, 10, 1449–1451. doi:10.1021/ol800224k
Return to citation in text: [1] -
Yan, S.; Gao, Y.; Xing, R.; Shen, Y.; Liu, Y.; Wu, P.; Wu, H. Tetrahedron 2008, 64, 6294–6299. doi:10.1016/j.tet.2008.04.100
Return to citation in text: [1] -
Kancharla, P. K.; Reddy, Y. S.; Dharuman, S.; Vankar, Y. D. J. Org. Chem. 2011, 76, 5832–5837. doi:10.1021/jo200475h
Return to citation in text: [1] -
Anderson, J. C.; Stepney, G. J.; Mills, M. R.; Horsfall, L. R.; Blake, A. J.; Lewis, W. J. Org. Chem. 2011, 76, 1961–1971. doi:10.1021/jo102408u
Return to citation in text: [1] [2] -
Anderson, J. C.; Blake, A. J.; Koovits, P. J.; Stepney, G. J. J. Org. Chem. 2012, 77, 4711–4724. doi:10.1021/jo300535h
Return to citation in text: [1] [2] -
Lucet, D.; Le Gall, T.; Mioskowski, C. Angew. Chem., Int. Ed. 1998, 37, 2580–2627. doi:10.1002/(SICI)1521-3773(19981016)37:19<2580::AID-ANIE2580>3.0.CO;2-L
Return to citation in text: [1] -
Ballini, R.; Petrini, M. Tetrahedron 2004, 60, 1017–1047. doi:10.1016/j.tet.2003.11.016
Return to citation in text: [1] -
Kizirian, J.-C. Chem. Rev. 2008, 108, 140–205. doi:10.1021/cr040107v
Return to citation in text: [1] -
Martjuga, M.; Shabashov, D.; Belyakov, S.; Liepinish, E.; Suna, E. J. Org. Chem. 2010, 75, 2357–2368. doi:10.1021/jo100173f
Return to citation in text: [1] -
Patrocinio, V. L.; Costa, P. R. R.; Correia, C. R. D. Synthesis 1994, 474–476. doi:10.1055/s-1994-25504
Return to citation in text: [1] -
Costa, J. S.; Dias, A. G.; Anholeto, A. L.; Monteiro, M. D.; Patrocinio, V. L.; Costa, P. R. R. J. Org. Chem. 1997, 62, 4002–4006. doi:10.1021/jo960788x
Return to citation in text: [1] -
Silva, P. C.; Costa, J. S.; Pereira, V. L. P. Synth. Commun. 2001, 31, 595–600. doi:10.1081/SCC-100000587
Return to citation in text: [1] -
Pinto, A. C.; Freitas, C. B. L.; Dias, A. G.; Pereira, V. L. P.; Tinant, B.; Declercq, J.-P.; Costa, P. R. R. Tetrahedron: Asymmetry 2002, 13, 1025–1031. doi:10.1016/S0957-4166(02)00230-6
Return to citation in text: [1] -
Costa, J. S.; Freire, B. S.; Moura, A. L. S.; Pereira, V. L. P. J. Braz. Chem. Soc. 2006, 17, 1229–1232. doi:10.1590/S0103-50532006000700006
Return to citation in text: [1] -
Pennaforte, E. V.; Costa, J. S.; Silva, C. A.; Saraiva, M. C.; Pereira, V. L. P. Lett. Org. Chem. 2009, 6, 110–114. doi:10.2174/157017809787582771
Return to citation in text: [1] -
Barreto, C. B., Jr.; Pereira, V. L. P. Tetrahedron Lett. 2009, 50, 6389–6392. doi:10.1016/j.tetlet.2009.08.087
Return to citation in text: [1] -
de Souza, R. O. M. A.; Fregadolli, P. H.; Gonçalves, K. M.; Sequeira, L. C.; Pereira, V. L. P.; Filho, L. C.; Esteves, P. M.; Vasconcellos, M. L. A. A.; Antunes, O. A. C. Lett. Org. Chem. 2006, 3, 936–939. doi:10.2174/157017806779467942
Return to citation in text: [1] [2] -
Reetz, M. T. Chem. Rev. 1999, 99, 1121–1162. doi:10.1021/cr980417b
Return to citation in text: [1] -
Reetz, M. T.; Drewes, M. W.; Schwickardi, R. Org. Synth. 2004, Coll. Vol. 10, 256–262.
Return to citation in text: [1] -
Beaulieu, P. L.; Wernic, D. J. Org. Chem. 1996, 61, 3635–3645. doi:10.1021/jo960109i
Return to citation in text: [1] -
O'Brien, P.; Warren, S. Tetrahedron Lett. 1996, 37, 4271–4274. doi:10.1016/0040-4039(96)00814-3
Return to citation in text: [1] -
Hanessian, S.; Devasthale, P. V. Tetrahedron Lett. 1996, 37, 987–990. doi:10.1016/0040-4039(95)02359-3
Return to citation in text: [1] -
Sinhababu, A. K.; Borchardt, R. T. Tetrahedron Lett. 1983, 24, 227–230. doi:10.1016/S0040-4039(00)81371-4
Return to citation in text: [1] -
Nising, C. F.; Bräse, S. Chem. Soc. Rev. 2008, 37, 1218–1228. doi:10.1039/B718357G
Return to citation in text: [1] -
Nising, C. F.; Bräse, S. Chem. Soc. Rev. 2012, 41, 988–999. doi:10.1039/C1CS15167C
Return to citation in text: [1] -
Beak, P.; Anderson, D. R.; Curtis, M. D.; Laumer, J. M.; Pippel, D. J.; Weisenburger, G. A. Acc. Chem. Res. 2000, 33, 715–727. doi:10.1021/ar000077s
Return to citation in text: [1] -
Paulsen, H.; Greve, W. Chem. Ber. 1974, 107, 3013–3019. doi:10.1002/cber.19741070923
Return to citation in text: [1] [2] [3] -
Sakakibara, T.; Sudoh, R. J. Org. Chem. 1977, 42, 1746–1750. doi:10.1021/jo00430a017
Return to citation in text: [1] [2] [3] -
Mangione, M. I.; Suárez, A. G.; Spanevello, R. A. Carbohydr. Res. 2005, 340, 149–153. doi:10.1016/j.carres.2004.10.018
Return to citation in text: [1] [2] -
Bernardi, L.; Fini, F.; Fochi, M.; Ricci, A. Synlett 2008, 1857–1861. doi:10.1055/s-2008-1078496
Return to citation in text: [1] [2] -
Anderson, J. C.; Blake, A. J.; Mills, M.; Ratcliffe, P. D. Org. Lett. 2008, 10, 4141–4143. doi:10.1021/ol801691c
Return to citation in text: [1] [2] -
Bernal, P.; Fernández, R.; Lassaletta, J. M. Chem.–Eur. J. 2010, 16, 7714–7718. doi:10.1002/chem.201001107
Return to citation in text: [1] [2] -
Yeom, C.-E.; Kim, M. J.; Kim, B. M. Tetrahedron 2007, 63, 904–909. doi:10.1016/j.tet.2006.11.037
Return to citation in text: [1] -
Bernasconi, C. F.; Renfrow, R. A.; Tia, P. R. J. Am. Chem. Soc. 1986, 108, 4541–4549. doi:10.1021/ja00275a047
Return to citation in text: [1] -
Gottlieb, L.; Hassner, A. J. Org. Chem. 1995, 60, 3759–3763. doi:10.1021/jo00117a030
Return to citation in text: [1] -
Hynes, P. S.; Stupple, P. A.; Dixon, D. J. Org. Lett. 2008, 10, 1389–1391. doi:10.1021/ol800108u
Return to citation in text: [1]
| 46. | Qian, C.; Takayuki, O.; Hasner, A. Synth. Commun. 2000, 30, 293–300. doi:10.1080/00397910008087321 |
| 75. | Nising, C. F.; Bräse, S. Chem. Soc. Rev. 2008, 37, 1218–1228. doi:10.1039/B718357G |
| 76. | Nising, C. F.; Bräse, S. Chem. Soc. Rev. 2012, 41, 988–999. doi:10.1039/C1CS15167C |
| 77. | Beak, P.; Anderson, D. R.; Curtis, M. D.; Laumer, J. M.; Pippel, D. J.; Weisenburger, G. A. Acc. Chem. Res. 2000, 33, 715–727. doi:10.1021/ar000077s |
| 78. | Paulsen, H.; Greve, W. Chem. Ber. 1974, 107, 3013–3019. doi:10.1002/cber.19741070923 |
| 79. | Sakakibara, T.; Sudoh, R. J. Org. Chem. 1977, 42, 1746–1750. doi:10.1021/jo00430a017 |
| 80. | Mangione, M. I.; Suárez, A. G.; Spanevello, R. A. Carbohydr. Res. 2005, 340, 149–153. doi:10.1016/j.carres.2004.10.018 |
| 81. | Bernardi, L.; Fini, F.; Fochi, M.; Ricci, A. Synlett 2008, 1857–1861. doi:10.1055/s-2008-1078496 |
| 82. | Anderson, J. C.; Blake, A. J.; Mills, M.; Ratcliffe, P. D. Org. Lett. 2008, 10, 4141–4143. doi:10.1021/ol801691c |
| 83. | Bernal, P.; Fernández, R.; Lassaletta, J. M. Chem.–Eur. J. 2010, 16, 7714–7718. doi:10.1002/chem.201001107 |
| 1. | Barrett, A. G. M. Chem. Soc. Rev. 1991, 20, 95–127. doi:10.1039/cs9912000095 |
| 2. | Ono, N. The Nitro Group in Organic Synthesis; Wiley-VCH: New York, 2001. doi:10.1002/0471224480 |
| 3. | Ballini, R.; Gabrielli, S.; Palmieri, A. Curr. Org. Chem. 2010, 14, 65–83. doi:10.2174/138527210790226429 |
| 4. | Parry, R.; Nishino, S.; Spain, J. Nat. Prod. Rep. 2011, 28, 152–167. doi:10.1039/c0np00024h |
| 19. | Amantini, D.; Fringuelli, F.; Piermatti, O.; Pizzo, F.; Zunino, E.; Vaccaro, L. J. Org. Chem. 2005, 70, 6526–6529. doi:10.1021/jo0507845 |
| 20. | Quiclet-Sire, B.; Zard, S. Z. Synthesis 2005, 3319–3326. doi:10.1055/s-2005-918463 |
| 21. | Zou, W.; Bhasin, M.; Vembaiyan, K.; Williams, D. T. Carbohydr. Res. 2009, 344, 1024–1027. doi:10.1016/j.carres.2009.03.007 |
| 22. | Arai, T.; Mishiro, A.; Yokoyama, N.; Suzuki, K.; Sato, H. J. Am. Chem. Soc. 2010, 132, 5338–5339. doi:10.1021/ja100265j |
| 52. | Fioravanti, S.; Pellacani, L.; Tardella, P. A.; Vergari, M. C. Org. Lett. 2008, 10, 1449–1451. doi:10.1021/ol800224k |
| 53. | Yan, S.; Gao, Y.; Xing, R.; Shen, Y.; Liu, Y.; Wu, P.; Wu, H. Tetrahedron 2008, 64, 6294–6299. doi:10.1016/j.tet.2008.04.100 |
| 54. | Kancharla, P. K.; Reddy, Y. S.; Dharuman, S.; Vankar, Y. D. J. Org. Chem. 2011, 76, 5832–5837. doi:10.1021/jo200475h |
| 8. | Robiette, R.; Tran, T.-V.; Cordi, A.; Tinant, B.; Marchand-Brynaert, J. Synthesis 2010, 3138–3142. doi:10.1055/s-0030-1258176 |
| 19. | Amantini, D.; Fringuelli, F.; Piermatti, O.; Pizzo, F.; Zunino, E.; Vaccaro, L. J. Org. Chem. 2005, 70, 6526–6529. doi:10.1021/jo0507845 |
| 20. | Quiclet-Sire, B.; Zard, S. Z. Synthesis 2005, 3319–3326. doi:10.1055/s-2005-918463 |
| 21. | Zou, W.; Bhasin, M.; Vembaiyan, K.; Williams, D. T. Carbohydr. Res. 2009, 344, 1024–1027. doi:10.1016/j.carres.2009.03.007 |
| 22. | Arai, T.; Mishiro, A.; Yokoyama, N.; Suzuki, K.; Sato, H. J. Am. Chem. Soc. 2010, 132, 5338–5339. doi:10.1021/ja100265j |
| 15. | Rastogi, N.; Namboothiri, I. N. N.; Cojocaru, M. Tetrahedron Lett. 2004, 45, 4745–4748. doi:10.1016/j.tetlet.2004.04.069 |
| 16. | Deb, I.; Shanbhag, P.; Mobin, S. M.; Namboothiri, I. N. N. Eur. J. Org. Chem. 2009, 4091–4101. doi:10.1002/ejoc.200900475 |
| 17. | Kuan, H.-H.; Reddy, R. J.; Chen, K. Tetrahedron 2010, 66, 9875–9879. doi:10.1016/j.tet.2010.10.061 |
| 18. | Bakthadoss, M.; Sivakumar, N.; Devaraj, A.; Sharada, D. S. Synthesis 2011, 2136–2146. doi:10.1055/s-0030-1260053 |
| 55. | Anderson, J. C.; Stepney, G. J.; Mills, M. R.; Horsfall, L. R.; Blake, A. J.; Lewis, W. J. Org. Chem. 2011, 76, 1961–1971. doi:10.1021/jo102408u |
| 56. | Anderson, J. C.; Blake, A. J.; Koovits, P. J.; Stepney, G. J. J. Org. Chem. 2012, 77, 4711–4724. doi:10.1021/jo300535h |
| 8. | Robiette, R.; Tran, T.-V.; Cordi, A.; Tinant, B.; Marchand-Brynaert, J. Synthesis 2010, 3138–3142. doi:10.1055/s-0030-1258176 |
| 11. | Poulsen, T. B.; Jørgensen, K. A. Chem. Rev. 2008, 108, 2903–2915. doi:10.1021/cr078372e |
| 12. | Trost, B. M.; Müller, C. J. Am. Chem. Soc. 2008, 130, 2438–2439. doi:10.1021/ja711080y |
| 13. | Yuan, Z.-L.; Lei, Z.-Y.; Shi, M. Tetrahedron: Asymmetry 2008, 19, 1339–1346. doi:10.1016/j.tetasy.2008.04.037 |
| 14. | Carmona, D.; Lamata, M. P.; Sánchez, A.; Viguri, F.; Oro, L. A. Tetrahedron: Asymmetry 2011, 22, 893–906. doi:10.1016/j.tetasy.2011.05.003 |
| 46. | Qian, C.; Takayuki, O.; Hasner, A. Synth. Commun. 2000, 30, 293–300. doi:10.1080/00397910008087321 |
| 47. | Hübner, J.; Liebscher, J.; Pätzel, M. Tetrahedron 2002, 58, 10485–10500. doi:10.1016/S0040-4020(02)01406-0 |
| 48. | Sakakibara, T.; Tachmori, Y.; Sudoh, R. Tetrahedron 2004, 40, 1533–1539. doi:10.1016/S0040-4020(01)91801-0 |
| 49. | Fioravanti, S.; Marchetti, F.; Pellacani, L.; Ranieri, L.; Tardella, P. A. Tetrahedron: Asymmetry 2008, 19, 231–236. doi:10.1016/j.tetasy.2007.12.007 |
| 50. | Lehnert, T.; Özüduru, G.; Grugel, H.; Albrecht, F.; Telligmann, S. M.; Boysen, M. M. K. Synthesis 2011, 2685–2708. doi:10.1055/s-0030-1260143 |
| 78. | Paulsen, H.; Greve, W. Chem. Ber. 1974, 107, 3013–3019. doi:10.1002/cber.19741070923 |
| 81. | Bernardi, L.; Fini, F.; Fochi, M.; Ricci, A. Synlett 2008, 1857–1861. doi:10.1055/s-2008-1078496 |
| 82. | Anderson, J. C.; Blake, A. J.; Mills, M.; Ratcliffe, P. D. Org. Lett. 2008, 10, 4141–4143. doi:10.1021/ol801691c |
| 83. | Bernal, P.; Fernández, R.; Lassaletta, J. M. Chem.–Eur. J. 2010, 16, 7714–7718. doi:10.1002/chem.201001107 |
| 5. | Wu, J.; Mampreian, D. M.; Hoveyda, A. H. J. Am. Chem. Soc. 2005, 127, 4584–4585. doi:10.1021/ja050800f |
| 6. | Rasappan, R.; Reiser, O. Eur. J. Org. Chem. 2009, 1305–1308. doi:10.1002/ejoc.200801263 |
| 7. | Muruganantham, R.; Namboothiri, I. J. Org. Chem. 2010, 75, 2197–2205. doi:10.1021/jo902595e |
| 8. | Robiette, R.; Tran, T.-V.; Cordi, A.; Tinant, B.; Marchand-Brynaert, J. Synthesis 2010, 3138–3142. doi:10.1055/s-0030-1258176 |
| 9. | Zhong, C.; Gautam, L. N. S.; Wang, D.; Akhmedov, N. G.; Petersen, J. L.; Shi, X. Tetrahedron 2011, 67, 4402–4411. doi:10.1016/j.tet.2011.01.085 |
| 10. | Rulev, A. Y. Russ. Chem. Rev. 2011, 80, 197–218. |
| 51. | Melton, J.; McMurry, J. E. J. Org. Chem. 1975, 40, 2138–2139. doi:10.1021/jo00902a033 |
| 15. | Rastogi, N.; Namboothiri, I. N. N.; Cojocaru, M. Tetrahedron Lett. 2004, 45, 4745–4748. doi:10.1016/j.tetlet.2004.04.069 |
| 16. | Deb, I.; Shanbhag, P.; Mobin, S. M.; Namboothiri, I. N. N. Eur. J. Org. Chem. 2009, 4091–4101. doi:10.1002/ejoc.200900475 |
| 17. | Kuan, H.-H.; Reddy, R. J.; Chen, K. Tetrahedron 2010, 66, 9875–9879. doi:10.1016/j.tet.2010.10.061 |
| 18. | Bakthadoss, M.; Sivakumar, N.; Devaraj, A.; Sharada, D. S. Synthesis 2011, 2136–2146. doi:10.1055/s-0030-1260053 |
| 68. | de Souza, R. O. M. A.; Fregadolli, P. H.; Gonçalves, K. M.; Sequeira, L. C.; Pereira, V. L. P.; Filho, L. C.; Esteves, P. M.; Vasconcellos, M. L. A. A.; Antunes, O. A. C. Lett. Org. Chem. 2006, 3, 936–939. doi:10.2174/157017806779467942 |
| 33. | Beller, M.; Bolm, C. Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, 2nd ed.; Wiley-VCH: Weinheim, 2004. doi:10.1002/9783527619405 |
| 34. | Jia, Y.-X.; Zhu, S.-F.; Yang, Y.; Zhou, Q.-L. J. Org. Chem. 2006, 71, 75–80. doi:10.1021/jo0516537 |
| 35. | Arai, T.; Yokoyama, N. Angew. Chem., Int. Ed. 2008, 47, 4989–4992. doi:10.1002/anie.200801373 |
| 36. | Gutnov, A. Eur. J. Org. Chem. 2008, 4547–4554. doi:10.1002/ejoc.200800541 |
| 37. | Zhang, W.-H.; Chien, S. W.; Hor, T. S. A. Coord. Chem. Rev. 2011, 255, 1991–2024. doi:10.1016/j.ccr.2011.05.018 |
| 42. | Shao, Z.; Zhang, H. Chem. Soc. Rev. 2009, 38, 2745–2755. doi:10.1039/b901258n |
| 43. | Zhong, C.; Shi, X. Eur. J. Org. Chem. 2010, 2999–3025. doi:10.1002/ejoc.201000004 |
| 44. | Alemán, J.; del Solar, V.; Martín-Santos, C.; Cubo, L.; Ranninger, C. N. J. Org. Chem. 2011, 76, 7287–7293. doi:10.1021/jo2013077 |
| 79. | Sakakibara, T.; Sudoh, R. J. Org. Chem. 1977, 42, 1746–1750. doi:10.1021/jo00430a017 |
| 32. | Denmark, S. E.; Schnute, M. E. J. Org. Chem. 1995, 60, 1013–1019. doi:10.1021/jo00109a037 |
| 45. | Hanessian, S.; Franco, J.; Larouche, B. Pure Appl. Chem. 1990, 62, 1887–1910. doi:10.1351/pac199062101887 |
| 80. | Mangione, M. I.; Suárez, A. G.; Spanevello, R. A. Carbohydr. Res. 2005, 340, 149–153. doi:10.1016/j.carres.2004.10.018 |
| 27. | Denmark, S. E.; Thorarensen, A. Chem. Rev. 1996, 96, 137–165. doi:10.1021/cr940277f |
| 28. | Fringuelli, F.; Matteucci, M.; Piermatti, O.; Pizzo, F.; Burla, M. C. J. Org. Chem. 2001, 66, 4661–4666. doi:10.1021/jo010182v |
| 29. | Denmark, S. E.; Baiazitov, R. Y. J. Org. Chem. 2006, 71, 593–605. doi:10.1021/jo052001l |
| 30. | Denmark, S. E.; Baiazitov, R. Y.; Nguyen, S. T. Tetrahedron 2009, 65, 6535–6548. doi:10.1016/j.tet.2009.05.060 |
| 31. | Korotaev, V. Y.; Sosnovskikh, V. Y.; Barabanov, M. A.; Yasnova, E. S.; Ezhikova, M. A.; Kodess, M. I.; Slepukhin, P. A. Tetrahedron 2010, 66, 1404–1409. doi:10.1016/j.tet.2009.11.094 |
| 78. | Paulsen, H.; Greve, W. Chem. Ber. 1974, 107, 3013–3019. doi:10.1002/cber.19741070923 |
| 23. | Kim, W. H.; Lee, J. H.; Danishefsky, S. J. J. Am. Chem. Soc. 2009, 131, 12576–12578. doi:10.1021/ja9058926 |
| 24. | Araújo, N.; Gil, M. V.; Román, E.; Serrano, J. A. Tetrahedron 2010, 66, 2664–2674. doi:10.1016/j.tet.2010.02.027 |
| 25. | Mangione, M. I.; Sarotti, A. M.; Suárez, A. G.; Spanevello, R. A. Carbohydr. Res. 2011, 346, 460–464. doi:10.1016/j.carres.2010.12.006 |
| 26. | Butt, N. A.; Moody, C. J. Org. Lett. 2011, 13, 2224–2227. doi:10.1021/ol200477s |
| 38. | Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r |
| 39. | List, B. Chem. Rev. 2007, 107, 5413–5415. doi:10.1021/cr078412e |
| 40. | Bertelsen, S.; Jørgensen, K. A. Chem. Soc. Rev. 2009, 38, 2178–2189. doi:10.1039/b903816g |
| 41. | Cheong, P. H.-Y.; Legault, C. Y.; Um, J. M.; Çelebi-Ölcüm, N.; Houk, K. N. Chem. Rev. 2011, 111, 5042–5137. doi:10.1021/cr100212h |
| 79. | Sakakibara, T.; Sudoh, R. J. Org. Chem. 1977, 42, 1746–1750. doi:10.1021/jo00430a017 |
| 55. | Anderson, J. C.; Stepney, G. J.; Mills, M. R.; Horsfall, L. R.; Blake, A. J.; Lewis, W. J. Org. Chem. 2011, 76, 1961–1971. doi:10.1021/jo102408u |
| 56. | Anderson, J. C.; Blake, A. J.; Koovits, P. J.; Stepney, G. J. J. Org. Chem. 2012, 77, 4711–4724. doi:10.1021/jo300535h |
| 57. | Lucet, D.; Le Gall, T.; Mioskowski, C. Angew. Chem., Int. Ed. 1998, 37, 2580–2627. doi:10.1002/(SICI)1521-3773(19981016)37:19<2580::AID-ANIE2580>3.0.CO;2-L |
| 31. | Korotaev, V. Y.; Sosnovskikh, V. Y.; Barabanov, M. A.; Yasnova, E. S.; Ezhikova, M. A.; Kodess, M. I.; Slepukhin, P. A. Tetrahedron 2010, 66, 1404–1409. doi:10.1016/j.tet.2009.11.094 |
| 32. | Denmark, S. E.; Schnute, M. E. J. Org. Chem. 1995, 60, 1013–1019. doi:10.1021/jo00109a037 |
| 33. | Beller, M.; Bolm, C. Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, 2nd ed.; Wiley-VCH: Weinheim, 2004. doi:10.1002/9783527619405 |
| 58. | Ballini, R.; Petrini, M. Tetrahedron 2004, 60, 1017–1047. doi:10.1016/j.tet.2003.11.016 |
| 84. | Yeom, C.-E.; Kim, M. J.; Kim, B. M. Tetrahedron 2007, 63, 904–909. doi:10.1016/j.tet.2006.11.037 |
| 15. | Rastogi, N.; Namboothiri, I. N. N.; Cojocaru, M. Tetrahedron Lett. 2004, 45, 4745–4748. doi:10.1016/j.tetlet.2004.04.069 |
| 85. | Bernasconi, C. F.; Renfrow, R. A.; Tia, P. R. J. Am. Chem. Soc. 1986, 108, 4541–4549. doi:10.1021/ja00275a047 |
| 86. | Gottlieb, L.; Hassner, A. J. Org. Chem. 1995, 60, 3759–3763. doi:10.1021/jo00117a030 |
| 73. | Hanessian, S.; Devasthale, P. V. Tetrahedron Lett. 1996, 37, 987–990. doi:10.1016/0040-4039(95)02359-3 |
| 74. | Sinhababu, A. K.; Borchardt, R. T. Tetrahedron Lett. 1983, 24, 227–230. doi:10.1016/S0040-4039(00)81371-4 |
| 71. | Beaulieu, P. L.; Wernic, D. J. Org. Chem. 1996, 61, 3635–3645. doi:10.1021/jo960109i |
| 72. | O'Brien, P.; Warren, S. Tetrahedron Lett. 1996, 37, 4271–4274. doi:10.1016/0040-4039(96)00814-3 |
| 45. | Hanessian, S.; Franco, J.; Larouche, B. Pure Appl. Chem. 1990, 62, 1887–1910. doi:10.1351/pac199062101887 |
| 69. | Reetz, M. T. Chem. Rev. 1999, 99, 1121–1162. doi:10.1021/cr980417b |
| 70. | Reetz, M. T.; Drewes, M. W.; Schwickardi, R. Org. Synth. 2004, Coll. Vol. 10, 256–262. |
| 59. | Kizirian, J.-C. Chem. Rev. 2008, 108, 140–205. doi:10.1021/cr040107v |
| 60. | Martjuga, M.; Shabashov, D.; Belyakov, S.; Liepinish, E.; Suna, E. J. Org. Chem. 2010, 75, 2357–2368. doi:10.1021/jo100173f |
| 87. | Hynes, P. S.; Stupple, P. A.; Dixon, D. J. Org. Lett. 2008, 10, 1389–1391. doi:10.1021/ol800108u |
| 61. | Patrocinio, V. L.; Costa, P. R. R.; Correia, C. R. D. Synthesis 1994, 474–476. doi:10.1055/s-1994-25504 |
| 62. | Costa, J. S.; Dias, A. G.; Anholeto, A. L.; Monteiro, M. D.; Patrocinio, V. L.; Costa, P. R. R. J. Org. Chem. 1997, 62, 4002–4006. doi:10.1021/jo960788x |
| 63. | Silva, P. C.; Costa, J. S.; Pereira, V. L. P. Synth. Commun. 2001, 31, 595–600. doi:10.1081/SCC-100000587 |
| 64. | Pinto, A. C.; Freitas, C. B. L.; Dias, A. G.; Pereira, V. L. P.; Tinant, B.; Declercq, J.-P.; Costa, P. R. R. Tetrahedron: Asymmetry 2002, 13, 1025–1031. doi:10.1016/S0957-4166(02)00230-6 |
| 65. | Costa, J. S.; Freire, B. S.; Moura, A. L. S.; Pereira, V. L. P. J. Braz. Chem. Soc. 2006, 17, 1229–1232. doi:10.1590/S0103-50532006000700006 |
| 66. | Pennaforte, E. V.; Costa, J. S.; Silva, C. A.; Saraiva, M. C.; Pereira, V. L. P. Lett. Org. Chem. 2009, 6, 110–114. doi:10.2174/157017809787582771 |
| 67. | Barreto, C. B., Jr.; Pereira, V. L. P. Tetrahedron Lett. 2009, 50, 6389–6392. doi:10.1016/j.tetlet.2009.08.087 |
| 68. | de Souza, R. O. M. A.; Fregadolli, P. H.; Gonçalves, K. M.; Sequeira, L. C.; Pereira, V. L. P.; Filho, L. C.; Esteves, P. M.; Vasconcellos, M. L. A. A.; Antunes, O. A. C. Lett. Org. Chem. 2006, 3, 936–939. doi:10.2174/157017806779467942 |
© 2013 Pereira et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)