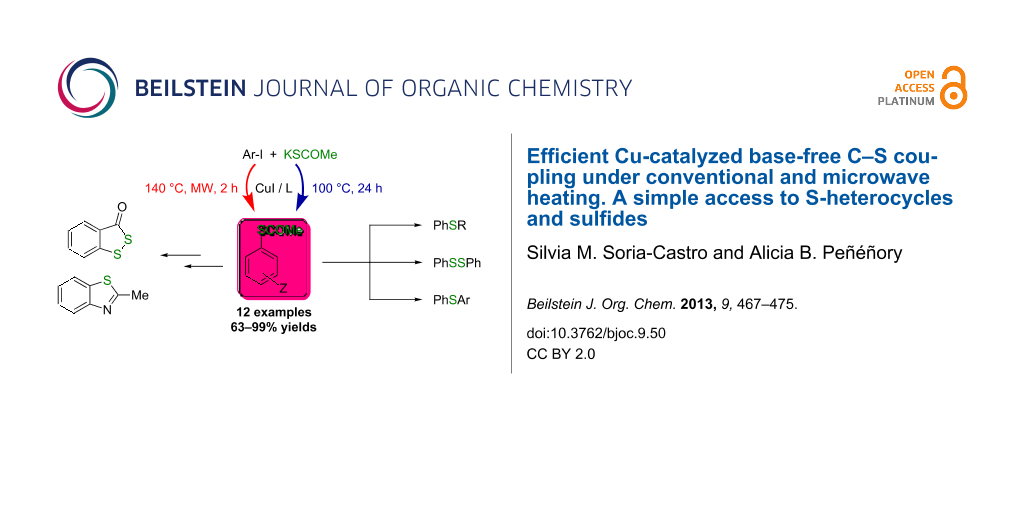
Abstract
S-aryl thioacetates can be prepared by reaction of inexpensive potassium thioacetate with both electron-rich and electron-poor aryl iodides under a base-free copper/ligand catalytic system. CuI as copper source affords S-aryl thioacetates in good to excellent yields, by using 1,10-phenanthroline as a ligand in toluene at 100 °C after 24 h. Under microwave irradiation the time was drastically reduced to 2 h. Both procedures are simple and involve a low-cost catalytic system. This methodology was also applied to the “one-pot” synthesis of target heterocycles, such as 3H-benzo[c][1,2]dithiol-3-one and 2-methylbenzothiazole, alkyl aryl sulfides, diaryl disulfides and asymmetric diaryl sulfides in good yields.

Graphical Abstract
Introduction
Aromatic sulfur compounds including sulfides and heterocycles are relevant in chemical, pharmaceutical and materials industries [1-3]. As a consequence, the development of new methodologies for CAr–S bond formation still represents a challenge in organic chemistry (Figure 1).
![[1860-5397-9-50-1]](/bjoc/content/figures/1860-5397-9-50-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of some pharmaceutically and chemically important derivatives.
Figure 1: Examples of some pharmaceutically and chemically important derivatives.
Nucleophilic substitution of aryl halides by a variety of sulfur anions, such as PhS−, 1-naphthylS−, S2−, thiourea anion (−SCNH(NH2)), thioacetate anion (MeCOS−) and thiobenzoate anion (PhCOS−), can be effectively achieved by photoinduced electron transfer reactions to form new C–S bonds [4,5]. We have previously described the reactivity of sulfur anions, such as S2− [6], −SCNH(NH2) [7,8], and MeCOS− [9], in photoinduced aromatic radical nucleophilic substitution (SRN1 mechanism), as a “one-pot” method for the synthesis of several sulfur aromatic compounds in moderate to good yields. A common feature of these reactions concerns the formation of arylthiolate anions as intermediates, offering a viable alternative for the introduction of sulfur functionalities in aryl compounds.
The formation of C–S bonds can also be accomplished in good to excellent yields by transition-metal catalysis [10,11], mainly using palladium [12], copper [13-16], nickel [17,18] and iron [19,20] salts. Aryl coupling reactions employing different palladium species as catalysts represent the most extensively studied approaches to form new CAr–S bonds [21-23]. Nevertheless, copper or iron-mediated coupling reactions have become a convenient alternative to the expensive Pd/ligand systems due to the lower cost of the former, its stability, and the ready availability of the ligands. Thus, different copper-salt-based catalytic systems have been found to be effective for the aryl coupling reactions of ArSH [24-29], RSO2Na [30], KSCN [31,32], NH2CSNH2 [33,34], MeCSNH2 [35], KSCSOEt [36,37], Na2S·9H2O [38,39] and sulfur [40,41] as well as the synthesis of diverse heterocycles containing sulfur atoms by intra- [42-44] and intermolecular [45-47] reactions.
Aryl thioesters are versatile intermediates for the synthesis of a variety of sulfur compounds including aryl thiols, aryl alkyl sulfides, aryl sulfenyl chlorides, aryl sulfonyl chlorides [48] and sulfonamides [49]. Thioacetate and thiobenzoate derivatives have been synthesized by the reactions of thioacetate and thiobenzoate anions with arenediazonium tetrafluoroborates [50]. However, this methodology implies moderate overall yields and the handling of usually unstable diazonium salts. More recently, S-aryl thioacetates have been obtained by the catalyst system [Pd2(dba)3-Xantphos] in 1,4-dioxane under microwave heating at 160 °C in good yields [51]. The synthesis of arylthiobenzoates by a CuI/1,10-phenanthroline, starting from thiobenzoic acid and iPr2NEt (DIPEA) in toluene after 24 h at 110 °C, has also been reported [52].
As part of our ongoing study on sulfur chemistry, we are interested in the one-pot synthesis of target heterocycles such as the Beaucage reagent and benzothiazole as well as sulfides by using Cu-catalysis to mediate the C–S bond formation. Herein, we report the reactions of KSCOMe (1) with aryl iodides under a Cu/ligand/base-free system to afford S-aryl thioacetates 2 as thiol surrogates in good to excellent yields, under conventional heating and microwave irradiation as an efficient access to a variety of sulfur compounds.
Results and Discussion
Initially, we tried PhCOSH with PhI using the catalytic systems of CuI/1,10-phenanthroline in toluene according to the method described by Sawada [52]. This methodology proved particularly sensitive to the nature and concentration of the base used in the deprotonation of the phenyl thiobenzoic acid due to an undesired hydrolysis of the thioester primary product. Thus, the reaction with a PhI/PhCOSH/Cs2CO3 ratio of 1:1.5:2 afforded Ph2S without traces of the thioester, and with a ratio of 1:1.5:1.5 the PhSCOPh was isolated in only 30% yield. Finally, the formation of PhSCOPh in our hands increased to 84% (quantified by GC) by using DIPEA as base with a PhI/PhCOSH/DIPEA ratio of 1:1.2:2 (Sawada’s conditions) [52] (Scheme 1).
![[1860-5397-9-50-i1]](/bjoc/content/inline/1860-5397-9-50-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of S-phenyl benzothioate.
Scheme 1: Synthesis of S-phenyl benzothioate.
Considering that the commercially available salt KSCOMe (1) is a solid, is easy to handle, is less odorous, and avoids the use of a relative expensive base with a lesser impact on the environment, as compared with the liquid PhCOSH, we continued our study on the synthesis of sulfides by using salt 1. The model reaction of PhI with KSCOMe was selected to determine the optimal conditions (solvent and temperature) starting with the effective CuI and 1,10-phenanthroline catalytic system previously employed (Scheme 2).
![[1860-5397-9-50-i2]](/bjoc/content/inline/1860-5397-9-50-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of S-phenyl thioacetate.
Scheme 2: Synthesis of S-phenyl thioacetate.
The use of the polar solvents DMF, MeCN and DMSO gave no conversion at all or yielded only a trace amount of S-phenyl thioacetate (2a) together with Ph2S and Ph2S2 from the in situ generation of benzenethiolate anion (Table 1, entries 1–3). In addition, no reaction in THF occurred after 48 h of heating under reflux, whereas 2a was formed in low yield when the reaction was performed in 1,4-dioxane, (Table 1, entries 4 and 5). Conversely, better results were obtained in nonpolar toluene at 100 °C affording 34% isolated yield of 2a after 24 h. This coupling reaction did not occur under air or in the absence of the ligand and was improved to 96% yield by using 10 and 20 mol % of CuI salt and ligand, respectively, (Table 1, entries 6–9). Similarly, a series of ligands usually employed in copper-catalyzed cross-coupling reactions was also screened, such as L-proline, benzotriazole, tetramethylethylenediamine (TMEDA), dimethylethylenediamine (DMEDA), and acetylacetone, under the optimized reaction conditions with 1,10-phenanthroline (Table 1, entry 9). These ligands, with different coordinating and structural properties, gave no positive results, and PhI was quantitatively recovered.
Table 1: Screening of solvents for the CuI-catalyzed reaction of potassium thioacetate (1) with iodobenzene.a
| entry | solvent | temp. [°C] | time [h] | 2a, yield (%)b |
|---|---|---|---|---|
| 1 | DMF | 80 | 24 | 0 |
| 2 | MeCN | 80 | 39 | <5c |
| 3 | DMSO | 100 | 24 | <5c |
| 4 | THF | 67 | 48 | 0 |
| 5 | Dioxane | 100 | 24 | 6 |
| 6 | Toluene | 100 | 24 | 34d |
| 7e | Toluene | 100 | 24 | 0 |
| 8f | Toluene | 100 | 24 | 0 |
| 9g | Toluene | 100 | 24 | 96 |
aReaction conditions: CuI (5 mol %, 0.025 mmol), 1,10-phenanthroline (10 mol %, 0.05 mmol), 1 (0.75 mmol), PhI (0.5 mmol), solvent (4 mL), conventional heating. bQuantified by GC by the internal standard method. cTogether with Ph2S and Ph2S2 as main products. dIsolated yield. eUnder an air atmosphere. fIn the absence of ligand. gCuI (10 mol %), 1,10-phenanthroline (20 mol %).
The effect of the Cu(I) and Cu(II) salts on the coupling reaction was also evaluated, and the results are summarized in Table 2. CuI and CuCl exhibited similar catalytic activity as did Cu(II) chloride dihydrate (Table 2, entries 1–3). There is general agreement that Cu(I) is the catalytic species in copper-catalyzed arylation of C, N or O-nucleophiles, which can be generated from Cu(II) or Cu(0) precursors by an in situ reduction or oxidation, respectively [53-55]. On the other hand, OTf− or OAc− counter ions decrease catalytic activity in Cu(II) salts, in comparison to chloride ions (Table 2, entries 4 and 5). Catalysis by CuO also affords moderate yield (Table 2, entry 6). Finally, in the absence of a copper source, no reaction occurred, supporting the participation of the copper species in the coupling reaction between thioacetate ion and PhI (Table 2, entry 7).
Table 2: Screening of copper sources for the copper-catalyzed reaction of potassium thioacetate (1) with iodobenzene.a
| entry | [Cu] | 2a, yield (%)b |
|---|---|---|
| 1 | CuI | 96 |
| 2 | CuCl | 93 |
| 3 | CuCl2·2H2O | 87 |
| 4 | Cu(OAc)2 | 29 |
| 5 | Cu(OTf)2 | 37 |
| 6 | CuO | 41 |
| 7 | — | 0 |
aReaction conditions: [Cu] (10 mol %, 0.05 mmol); 1,10-phenanthroline (20 mol %, 0.1 mmol), 1 (0.75 mmol), PhI (0.5 mmol), toluene (4 mL), 100 °C (conventional heating), 24 h. bQuantified by GC by the internal standard method.
A variety of commercially available aryl iodides were subjected to the optimized reaction conditions (Table 2, entry 1) to determine the scope of this catalytic system for the synthesis of S-aryl thioacetates 2, and the results are summarized in Table 3.
Table 3: Copper-catalyzed reaction of aryl iodides with potassium thioacetate (1).a
| entry | aryl iodide | product | 2 yield (%)b method | |
|---|---|---|---|---|
| A | B | |||
| 1 | PhI |
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-50-i9.svg?max-width=637&scale=1.0)
|
96 (80) | 92 (77) |
| 2 | p-MeC6H4I |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-50-i10.svg?max-width=637&scale=1.0)
|
96 (84) | |
| 3 | p-MeOC6H4I |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-50-i11.svg?max-width=637&scale=1.0)
|
94 (74) | 74 (69) |
| 4 | p-MeCONHC6H4I |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-50-i12.svg?max-width=637&scale=1.0)
|
(72) | |
| 5 | p-MeCOC6H4I |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-50-i13.svg?max-width=637&scale=1.0)
|
(89) | 79 (69) |
| 6 | p-NO2C6H4I |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-50-i14.svg?max-width=637&scale=1.0)
|
(73) | |
| 7 | p-CNC6H4I |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-50-i15.svg?max-width=637&scale=1.0)
|
73 | 64 (58) |
| 8 | o-MeC6H4I |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-50-i16.svg?max-width=637&scale=1.0)
|
84 (71) | |
| 9 | o-MeOC6H4I |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-50-i17.svg?max-width=637&scale=1.0)
|
(76) | 75 (67) |
| 10 | 3-C5NH4I |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-50-i18.svg?max-width=637&scale=1.0)
|
(82) | |
| 11 | 1-C10H7I |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-50-i19.svg?max-width=637&scale=1.0)
|
(75) | |
| 12 | p-C6H4I2 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-50-i20.svg?max-width=637&scale=1.0)
|
(63) | 99 (70) |
| 13 | PhBr |
|
— | — |
aReaction conditions: CuI (10 mol %, 0.05 mmol); 1,10-phenanthroline (20 mol %, 0.1 mmol), 1 (0.75 mmol), ArI (0.5 mmol); method A: toluene (4 mL), 100 °C (conventional heating), 24 h; method B: toluene (2 mL), microwave irradiation at 25 W, temperature between 110 °C and 140 °C, 2 h. bQuantified by GC by the internal standard method and isolated yield between parentheses. cArI2:1 ratio of 1:2.
As described in Table 3, the coupling of thioacetate with aryl iodides bearing electron-withdrawing (EWGs) and electron-donating (EDGs) groups proceeded in good to excellent yields (63–96%). For instance, the para-methoxy and para-acetyl phenyl iodides afforded thioacetates 2c and 2e in 74% and 89% isolated yield, respectively (Table 3, entries 3 and 5). In the absence of the catalytic system CuI/1,10-phenanthroline, the aryl iodides bearing the para-acetyl, para-nitro and para-cyano substituents, did not react with potassium thioacetate (1) after 24 h in toluene at 110 °C. These observations rule out the possibility of an aromatic nucleophilic substitution to account for the obtained results.
Furthermore, sterically hindered ortho-substituted aryl iodides afforded good yields of the coupling products 2 (Table 3, entries 8 and 9). For example, the ortho- and para-methoxy aryl iodides rendered comparable isolated yields of thioacetate derivatives 2c and 2i (Table 3, entries 3 and 9). The reaction between 4-iodoaniline and 1 under CuI/1,10-phenanthroline catalysis afforded quantitatively the corresponding 4-iodoacetanilide as the only product. The acetylation of the amino group competes effectively with the cross-coupling reaction. A similar acetylation reaction was also observed with 2-iodophenol. In consequence, protected amino and hydroxy groups were required for the copper-mediated C–S bond formation to proceed properly (Table 3, entries 3, 4 and 9).
This copper-catalyzed coupling reaction was also applied to heteroaromatic and other aromatics such as 3-iodopyridine and 1-naphthyl iodide, affording thioacetate derivatives 3 and 4 in good isolated yields. Interestingly, the reaction of 1,4-diiodobenzene with thioacetate rendered the corresponding disubstituted product 5, a straightforward precursor of 1,4-benzenedithiol, in high isolated yield. As a result, the simple methodology here presented could be an attractive alternative for the synthesis of polysulfides and other sulfur relevant materials (Table 3, entries 10–12).
In a comparative fashion we have also performed these reactions under microwave irradiation in order to improve conversion times and yields (Table 3, Method B). These reactions occur in only 2 h with yields comparable to those under conventional heating, for EWGs and EDGs (Table 3, entries 1, 3, 5, 7, 9 and 12). Bromobenzene was unreactive towards 1 under both conventional and microwave heating (Table 3, entry 13), this Cu-catalyzed C–S coupling reaction being highly chemoselective for aryl iodides.
In comparison with the already reported procedure for the preparation of S-aryl thioacetates by Pd catalysis [51,56], the methodology herein described has the advantages of using a lower-cost copper salt and a stable and accessible ligand. Furthermore, the use of a solid air-stable KSCOMe salt instead of a highly inflammable liquid and corrosive PhCOSH and DIPEA [52] offered environmental advantages, due to the absence of any additional base and the ease of handling. Moreover the use of microwave irradiation with concomitant decrease of the reaction times to only 2 h is noteworthy.
Afterwards we applied this procedure to the synthesis of heterocycles by the cross-coupling reaction of suitable o-substituted aryl iodides with KSCOMe. The reduced form of the Beaucage reagent 6, a sulfurizing reagent used for the synthesis of phosphorothioate oligonucleotides [57], was synthesized by the reaction of 2-mercaptobenzoic acid (7) with KSCOMe (1), probably through the intermediate 8 (Scheme 3) [58]. We envisioned that compound 6 could be easily achieved by a cascade process starting with the copper-mediated reaction of 2-iodobenzoic acid (9) with salt 1 to yield the thioacetate precursor of 7 (Scheme 4).
![[1860-5397-9-50-i3]](/bjoc/content/inline/1860-5397-9-50-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 6 by reaction between 7 and 1.
Scheme 3: Synthesis of 6 by reaction between 7 and 1.
![[1860-5397-9-50-i4]](/bjoc/content/inline/1860-5397-9-50-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of 2-iodobenzoic acid (9) with 1.
Scheme 4: Reaction of 2-iodobenzoic acid (9) with 1.
When a toluene solution of 9 and 1 in a ratio of 1:1.5 was allowed to react in the presence of CuI/1,10-phenanthroline, a mixture of 2-mercaptobenzoic acid (7) and 3H-benzo[c][1,2]dithiol-3-one (6) was obtained in 10% and 27% isolated yields, respectively (Scheme 4).
Compounds 7 and 6 derive from the previous C–S coupling reaction. Thus, the expected 2-(acetylthio)benzoate (10) undergoes a series of acyl transfers, and therefore, thioanhydride 12, and consecutively compound 8, are formed. Irreversible oxidation of the latter finally affords heterocycle 6. The free thiol 7 can be also generated by acyl transfer from 2-mercaptoanhydride 11 (Scheme 5).
![[1860-5397-9-50-i5]](/bjoc/content/inline/1860-5397-9-50-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Suggested one-pot synthesis of heterocycle 6.
Scheme 5: Suggested one-pot synthesis of heterocycle 6.
By optimizing the reaction conditions, it was possible to improve the yield of 6. Hence, the copper-catalyzed reaction between iodide 9 and 1 in a ratio of 1:2 rendered only heterocycle 6 (53% isolated yield) in a one-pot procedure. (Increasing the ratio from 1:2 to 1:3 did not increase the isolated yield of 6). A possible mechanism to account for this involves a cascade of reactions starting with a cross-coupling and followed by consecutive acyl transfers (Scheme 5).
We also explored the possibility of building a benzothiazole ring by copper-catalyzed reaction of N-(2-iodophenyl)acetamide (13) with 1. Thus, together with benzothiazole 14, the reaction of 13 and 1 in a 1:1.5 ratio, in the presence of CuI/1,10-phenanthroline after 24 h of conventional heating, afforded low amounts of the substituted thioacetate derivative S-(2-acetoamidophenyl)thioacetate (15) and N-(2-mercaptophenyl)acetamide (16) or S-(2-aminophenyl)thioacetate (17) without cyclization, as detected by GC–MS spectrometry (Scheme 6).
![[1860-5397-9-50-i6]](/bjoc/content/inline/1860-5397-9-50-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reaction of N-(2-iodophenyl)acetamide (13) with 1.
Scheme 6: Reaction of N-(2-iodophenyl)acetamide (13) with 1.
In order to accelerate the ring-closure reaction, a mixture of 13 and 1 in a 1:1.5 ratio was heated for 2 h under microwave irradiation, and 2-methylbenzothiazole (14) was isolated from the reaction mixture in 50% as the only reaction product (Scheme 7). Increasing the amount of 1 (13:1 in a 1:3 ratio) does not significantly increase the yield of 14 (57%).
![[1860-5397-9-50-i7]](/bjoc/content/inline/1860-5397-9-50-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of 2-methylbenzothiazole (14).
Scheme 7: Synthesis of 2-methylbenzothiazole (14).
This methodology was finally applied to the one-pot three-step synthesis of alkyl aryl sulfides, diaryldisulfides and asymmetric diaryl sulfides. Thus, without isolation, the phenyl thioacetate produced in good yield by the copper-catalyzed reaction of PhI with thioacetate anion 1 was hydrolyzed to the benzene thiolate anion by the addition of 2 equiv of KOt-Bu. Different chemical transformations are possible from the arene thiolate ions formed; for example, oxidation to the diaryl disulfide, subsequent nucleophilic substitution reaction with alkyl halides to afford alkyl aryl sulfides, or a second copper-catalyzed reaction with a different aryl iodide to yield asymmetric diaryl sulfides. With this strategy we synthesized different sulfur compounds. Accordingly, a solution of benzene thiolate obtained from this methodology affords bis(phenyl)disulfide (51%) after oxidation by KI/I2, by subsequent substitution reactions with methyl iodide and benzyl bromide, methyl phenyl sulfide and benzyl methyl sulfide in 81% and 66% yields, respectively, and 4-anisyl phenyl sulfide (4-AnSPh) in 85% yield (quantified by 1H NMR) by a second copper-catalyzed reaction (Scheme 8).
![[1860-5397-9-50-i8]](/bjoc/content/inline/1860-5397-9-50-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: One-pot three-step synthesis of sulfides. (a) KI/I2; (b) BrCH2Ph; (c) MeI; (d) 4-AnI, CuI/1,10-phenanthroline (10 and 20 mol %, respectively), 100 °C, 24 h.
Scheme 8: One-pot three-step synthesis of sulfides. (a) KI/I2; (b) BrCH2Ph; (c) MeI; (d) 4-AnI, CuI/1,10-phen...
Conclusion
We have developed a simple, versatile, efficient and economically attractive procedure for the synthesis of sulfur heterocycles and a variety of sulfides with good yields. This one-pot methodology involves a cascade of reactions starting with a C–S bond formation by copper catalysis and followed by consecutive acyl transfers, condensation, nucleophilic substitution, oxidation or a second copper cross-coupling of the arene thiolate anion intermediates from S-aryl thioacetate hydrolysis. These thioacetates are obtained by copper-catalyzed reaction of commercial potassium thioacetate with aryl iodides under microwave irradiation. Electron-donor and electron-acceptor substituents are tolerated, and polysubstitution can also be successfully accomplished.
Experimental
Representative experimental procedures for the Cu-catalyzed base-free C–S coupling
Method A: The reactions were carried out in a 10 mL two-necked Schlenk tube, equipped with a nitrogen gas inlet, a condenser and a magnetic stirrer. The tube was dried under vacuum, filled with nitrogen and then charged with dried toluene (4.0 mL). ArI (0.5 mmol), CuI (10 mol %), 1,10-phenanthroline (20 mol %) and finally potassium thioacetate (1) (0.75 mmol) were added to the degassed solvent under nitrogen and stirred at 100 °C for 24 h. The reaction mixture was cooled to room temperature. Diethylether (20 mL) and water (20 mL) were added and the mixture was stirred. The organic layer was separated and the aqueous layer was extracted with diethylether (2 × 20 mL). The combined organic extract was dried over Na2SO4, and the products were isolated by radial chromatography from the crude reaction mixture or quantified by GC by using the internal standard method. The identity of all the products was confirmed by 1H and 13C NMR and MS spectrometry. All the thioacetate compounds prepared are known, and their data are in good agreement with those reported.
Method B: The reactions were carried out in a 10 mL glass tube, filled with nitrogen and then charged with dried toluene (2 mL). ArI (0.5 mmol), CuI (10 mol %), 1,10-phenanthroline (20 mol %) and finally potassium thioacetate (1) (0.75 mmol) were added to the degassed solvent under nitrogen. Then, the tube was sealed with a rubber cap and heated to 110–140 °C for 2 h under microwave irradiation (Fixed Power, 25 W) by using the SPS method. This method implies irradiation at 25 W to bring the reaction mixture to 140 °C, then cycling of the power on and off for the remaining run time (2 h) as the temperature varies from 110–140 °C. After completion of the experiment, the vessel was cooled to room temperature before removal from the microwave cavity, and then opened to the atmosphere. The work-up of the reaction was similar to that of Method A.
Representative procedure for the one-pot three-step synthesis of alkyl aryl sulfides, diaryl disulfides and asymmetric diaryl sulfides
The reactions were carried out by using method A. After 24 h at 100 °C, KOt-Bu (1 mmol, 2 equiv) was added to the reaction mixture and stirred for 10 min. The corresponding alkyl halide (0.75 mmol, 1.5 equiv) or KI/I2 (1.5 mmol/0.51 mmol, 3/1.02 equiv) was then added and stirred for 20 min or 24 h, respectively. The work-up of the reactions was similar to that of Method A.
For the synthesis of the asymmetric diaryl sulfide, after hydrolysis of the thioester, a second addition of CuI/1,10-phenanthroline (10 and 20 mol %, respectively) was required, together with the new aryl iodide (1 equiv). After stirring for 24 h at 100 °C, the work-up of the reaction was similar to that of Method A.
Supporting Information
| Supporting Information File 1: Experimental details, characterization data and spectra (1H, 13C NMR, and HSQC or HMBC as consigned) for all the products (2a–i, 3–7, and 14). | ||
| Format: PDF | Size: 2.8 MB | Download |
References
-
Liu, G.; Huth, J. R.; Olejniczak, E. T.; Mendoza, R.; DeVries, P.; Leitza, S.; Reilly, E. B.; Okasinski, G. F.; Fesik, S. W.; von Geldern, T. W. J. Med. Chem. 2001, 44, 1202–1210. doi:10.1021/jm000503f
Return to citation in text: [1] -
You, N.-H.; Higashihara, T.; Oishi, Y.; Ando, S.; Ueda, M. Macromolecules 2010, 43, 4613–4615. doi:10.1021/ma100448d
Return to citation in text: [1] -
Marom, H.; Popowski, Y.; Antonov, S.; Gozin, M. Org. Lett. 2011, 13, 5532–5535. doi:10.1021/ol2022627
Return to citation in text: [1] -
Rossi, R. A.; Pierini, A. B.; Peñéñory, A. B. Chem. Rev. 2003, 103, 71–168. doi:10.1021/cr960134o
Return to citation in text: [1] -
Peñéñory, A. B.; Argüello, J. E. In Handbook of Synthetic Photochemistry; Albini, A.; Fagnoni, M., Eds.; Wiley-VCH Verlag GmbH &Co: Weinheim, 2010; pp 319–346.
Return to citation in text: [1] -
Argüello, J. E.; Schmidt, L. C.; Peñéñory, A. B. ARKIVOC 2003, No. x, 411–419.
Return to citation in text: [1] -
Argüello, J. E.; Schmidt, L. C.; Peñéñory, A. B. Org. Lett. 2003, 5, 4133–4136. doi:10.1021/ol035545n
Return to citation in text: [1] -
Argüello, J. E.; Schmidt, L. C.; Peñéñory, A. B. J. Org. Chem. 2007, 72, 2936–2944. doi:10.1021/jo062569+
Return to citation in text: [1] -
Schmidt, L. C.; Rey, V.; Peñéñory, A. B. Eur. J. Org. Chem. 2006, 2210–2214. doi:10.1002/ejoc.200500955
Return to citation in text: [1] -
Kondo, T.; Mitsudo, T.-a. Chem. Rev. 2000, 100, 3205–3220. doi:10.1021/cr9902749
Return to citation in text: [1] -
Beletskaya, I. P.; Ananikov, V. P. Chem. Rev. 2011, 111, 1596–1636. doi:10.1021/cr100347k
Return to citation in text: [1] -
Hartwig, J. F. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E.-I., Ed.; Wiley-Interscience: New York, 2002; Vol. 1, pp 1051–1096.
Return to citation in text: [1] -
Ley, S. V.; Thomas, A. W. Angew. Chem., Int. Ed. 2003, 42, 5400–5449. doi:10.1002/anie.200300594
Return to citation in text: [1] -
Beletskaya, I. P.; Cheprakov, A. V. Coord. Chem. Rev. 2004, 248, 2337–2364. doi:10.1016/j.ccr.2004.09.014
Return to citation in text: [1] -
Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2008, 47, 3096–3099. doi:10.1002/anie.200703209
Return to citation in text: [1] -
Qiao, J. X.; Lam, P. Y. S. Synthesis 2011, 829–856. doi:10.1055/s-0030-1258379
Return to citation in text: [1] -
Yoon, H.-J.; Choi, J.-W.; Kang, H.; Kang, T.; Lee, S.-M.; Jun, B.-H.; Lee, Y.-S. Synlett 2010, 2518–2522. doi:10.1055/s-0030-1258545
Return to citation in text: [1] -
Jammi, S.; Barua, P.; Rout, L.; Saha, P.; Punniyamurthy, T. Tetrahedron Lett. 2008, 49, 1484–1487. doi:10.1016/j.tetlet.2007.12.118
Return to citation in text: [1] -
Akkilagunta, V. K.; Reddy, V. P.; Rao, K. R. Synlett 2010, 1260–1264. doi:10.1055/s-0029-1219801
Return to citation in text: [1] -
Correa, A.; Carril, M.; Bolm, C. Angew. Chem. 2008, 120, 2922–2925. doi:10.1002/ange.200705668
Return to citation in text: [1] -
Wang, L.; Zhou, W.-Y.; Chen, S.-C.; He, M.-Y.; Chen, Q. Adv. Synth. Catal. 2012, 354, 839–845. doi:10.1002/adsc.201100788
Return to citation in text: [1] -
Fu, C.-F.; Liu, Y.-H.; Peng, S.-M.; Liu, S.-T. Tetrahedron 2010, 66, 2119–2122. doi:10.1016/j.tet.2010.01.081
Return to citation in text: [1] -
Fernández-Rodríguez, M. A.; Hartwig, J. F. J. Org. Chem. 2009, 74, 1663–1672. doi:10.1021/jo802594d
Return to citation in text: [1] -
Kwong, F. Y.; Buchwald, S. L. Org. Lett. 2002, 4, 3517–3520. doi:10.1021/ol0266673
Return to citation in text: [1] -
Bates, C. G.; Gujadhur, R. K.; Venkataraman, D. Org. Lett. 2002, 4, 2803–2806. doi:10.1021/ol0264105
Return to citation in text: [1] -
Sperotto, E.; van Klink, G. P. M.; de Vries, J. G.; van Koten, G. J. Org. Chem. 2008, 73, 5625–5628. doi:10.1021/jo800491k
Return to citation in text: [1] -
Herrero, M. T.; SanMartín, R.; Domínguez, E. Tetrahedron 2009, 65, 1500–1503. doi:10.1016/j.tet.2008.11.062
Return to citation in text: [1] -
Jammi, S.; Sakthivel, S.; Rout, L.; Mukherjee, T.; Mandal, S.; Mitra, R.; Saha, P.; Punniyamurthy, T. J. Org. Chem. 2009, 74, 1971–1976. doi:10.1021/jo8024253
Return to citation in text: [1] -
Feng, Y.-S.; Qi, H.-X.; Wang, W.-C.; Liang, Y.-F.; Xu, H.-J. Tetrahedron Lett. 2012, 53, 2914–2917. doi:10.1016/j.tetlet.2012.04.004
Return to citation in text: [1] -
Zhu, W.; Ma, D. J. Org. Chem. 2005, 70, 2696–2700. doi:10.1021/jo047758b
Return to citation in text: [1] -
Ke, F.; Qu, Y.; Jiang, Z.; Li, Z.; Wu, D.; Zhou, X. Org. Lett. 2011, 13, 454–457. doi:10.1021/ol102784c
Return to citation in text: [1] -
Reddy, K. H. V.; Reddy, V. P.; Kumar, A. A.; Kranthi, G.; Nageswar, Y. V. D. Beilstein J. Org. Chem. 2011, 7, 886–891. doi:10.3762/bjoc.7.101
Return to citation in text: [1] -
Reddy, K. H. V.; Reddy, V. P.; Shankar, J.; Madhav, B.; Kumar, B. S. P. A.; Nageswar, Y. V. D. Tetrahedron Lett. 2011, 52, 2679–2682. doi:10.1016/j.tetlet.2011.03.070
Return to citation in text: [1] -
Firouzabadi, H.; Iranpoor, N.; Gholinejad, M. Adv. Synth. Catal. 2010, 352, 119–124. doi:10.1002/adsc.200900671
Return to citation in text: [1] -
Tao, C.; Lv, A.; Zhao, N.; Yang, S.; Liu, X.; Zhou, J.; Liu, W.; Zhao, J. Synlett 2011, 134–138. doi:10.1055/s-0030-1259079
Return to citation in text: [1] -
Akkilagunta, V. K.; Kakulapati, R. R. J. Org. Chem. 2011, 76, 6819–6824. doi:10.1021/jo200793k
Return to citation in text: [1] -
Prasad, D. J. C.; Sekar, G. Org. Lett. 2011, 13, 1008–1011. doi:10.1021/ol103041s
Return to citation in text: [1] -
Ma, D.; Xie, S.; Xue, P.; Zhang, X.; Dong, J.; Jiang, Y. Angew. Chem., Int. Ed. 2009, 48, 4222–4225. doi:10.1002/anie.200900486
Return to citation in text: [1] -
Li, Y.; Nie, C.; Wang, H.; Li, X.; Verpoort, F.; Duan, C. Eur. J. Org. Chem. 2011, 7331–7338. doi:10.1002/ejoc.201101121
Return to citation in text: [1] -
Xu, H.-J.; Liang, Y.-F.; Cai, Z.-Y.; Qi, H.-X.; Yang, C.-Y.; Feng, Y.-S. J. Org. Chem. 2011, 76, 2296–2300. doi:10.1021/jo102506x
Return to citation in text: [1] -
Bhakuni, B. S.; Balkrishna, S. J.; Kumar, A.; Kumar, S. Tetrahedron Lett. 2012, 53, 1354–1357. doi:10.1016/j.tetlet.2012.01.003
Return to citation in text: [1] -
Evindar, G.; Batey, R. A. J. Org. Chem. 2006, 71, 1802–1808. doi:10.1021/jo051927q
Return to citation in text: [1] -
Ma, H. C.; Jiang, X. Z. Synlett 2008, 1335–1340. doi:10.1055/s-2008-1072764
Return to citation in text: [1] -
Murru, S.; Ghosh, H.; Sahoo, S. K.; Patel, B. K. Org. Lett. 2009, 11, 4254–4257. doi:10.1021/ol9017535
Return to citation in text: [1] -
Ma, D.; Geng, Q.; Zhang, H.; Jiang, Y. Angew. Chem., Int. Ed. 2010, 49, 1291–1294. doi:10.1002/anie.200905646
Return to citation in text: [1] -
Ma, D.; Lu, X.; Shi, L.; Zhang, H.; Jiang, Y.; Liu, X. Angew. Chem., Int. Ed. 2011, 50, 1118–1121. doi:10.1002/anie.201005787
Return to citation in text: [1] -
Li, J.; Zhang, Y.; Jiang, Y.; Ma, D. Tetrahedron Lett. 2012, 53, 2511–2513. doi:10.1016/j.tetlet.2012.03.006
Return to citation in text: [1] -
Nishiguchi, A.; Maeda, K.; Miki, S. Synthesis 2006, 4131–4134. doi:10.1055/s-2006-950353
Return to citation in text: [1] -
Ho, D. K. H.; Chan, L.; Hooper, A.; Brennan, P. E. Tetrahedron Lett. 2011, 52, 820–823. doi:10.1016/j.tetlet.2010.12.050
Return to citation in text: [1] -
Petrillo, G.; Novi, M.; Garbarino, G.; Filiberti, M. Tetrahedron 1989, 45, 7411–7420. doi:10.1016/S0040-4020(01)89203-6
Return to citation in text: [1] -
Lai, C.; Backes, B. J. Tetrahedron Lett. 2007, 48, 3033–3037. doi:10.1016/j.tetlet.2007.02.128
Return to citation in text: [1] [2] -
Sawada, N.; Itoh, T.; Yasuda, N. Tetrahedron Lett. 2006, 47, 6595–6597. doi:10.1016/j.tetlet.2006.07.008
Return to citation in text: [1] [2] [3] [4] -
Ouali, A.; Taillefer, M.; Spindler, J.-F.; Jutand, A. Organometallics 2007, 26, 65–74. doi:10.1021/om060706n
Return to citation in text: [1] -
Jones, G. O.; Liu, P.; Houk, K. N.; Buchwald, S. L. J. Am. Chem. Soc. 2010, 132, 6205–6213. doi:10.1021/ja100739h
Return to citation in text: [1] -
Huang, Z.; Hartwig, J. F. Angew. Chem., Int. Ed. 2012, 51, 1028–1032. doi:10.1002/anie.201106719
Return to citation in text: [1] -
General reaction conditions: Potassium thioacetate (1) (1 equiv), Pd2(dba)2, (2.5 mol %), Xantphos (5 mol %), i-Pr2NEt (2 equiv), 1,4-dioxane, microwave 160 °C, 25 min, [52].
Return to citation in text: [1] -
Iyer, R. P.; Egan, W.; Regan, J. B.; Beaucage, S. L. J. Am. Chem. Soc. 1990, 112, 1253–1254. doi:10.1021/ja00159a059
Return to citation in text: [1] -
Zheng, J.; Liu, X.; Quan, Q.; Shin, Y.-J.; Sun, D.; Lu, Y. Org. Biomol. Chem. 2010, 1293–1295. doi:10.1039/b926217b
Return to citation in text: [1]
| 52. | Sawada, N.; Itoh, T.; Yasuda, N. Tetrahedron Lett. 2006, 47, 6595–6597. doi:10.1016/j.tetlet.2006.07.008 |
| 53. | Ouali, A.; Taillefer, M.; Spindler, J.-F.; Jutand, A. Organometallics 2007, 26, 65–74. doi:10.1021/om060706n |
| 54. | Jones, G. O.; Liu, P.; Houk, K. N.; Buchwald, S. L. J. Am. Chem. Soc. 2010, 132, 6205–6213. doi:10.1021/ja100739h |
| 55. | Huang, Z.; Hartwig, J. F. Angew. Chem., Int. Ed. 2012, 51, 1028–1032. doi:10.1002/anie.201106719 |
| 51. | Lai, C.; Backes, B. J. Tetrahedron Lett. 2007, 48, 3033–3037. doi:10.1016/j.tetlet.2007.02.128 |
| 56. | General reaction conditions: Potassium thioacetate (1) (1 equiv), Pd2(dba)2, (2.5 mol %), Xantphos (5 mol %), i-Pr2NEt (2 equiv), 1,4-dioxane, microwave 160 °C, 25 min, [52]. |
| 1. | Liu, G.; Huth, J. R.; Olejniczak, E. T.; Mendoza, R.; DeVries, P.; Leitza, S.; Reilly, E. B.; Okasinski, G. F.; Fesik, S. W.; von Geldern, T. W. J. Med. Chem. 2001, 44, 1202–1210. doi:10.1021/jm000503f |
| 2. | You, N.-H.; Higashihara, T.; Oishi, Y.; Ando, S.; Ueda, M. Macromolecules 2010, 43, 4613–4615. doi:10.1021/ma100448d |
| 3. | Marom, H.; Popowski, Y.; Antonov, S.; Gozin, M. Org. Lett. 2011, 13, 5532–5535. doi:10.1021/ol2022627 |
| 9. | Schmidt, L. C.; Rey, V.; Peñéñory, A. B. Eur. J. Org. Chem. 2006, 2210–2214. doi:10.1002/ejoc.200500955 |
| 33. | Reddy, K. H. V.; Reddy, V. P.; Shankar, J.; Madhav, B.; Kumar, B. S. P. A.; Nageswar, Y. V. D. Tetrahedron Lett. 2011, 52, 2679–2682. doi:10.1016/j.tetlet.2011.03.070 |
| 34. | Firouzabadi, H.; Iranpoor, N.; Gholinejad, M. Adv. Synth. Catal. 2010, 352, 119–124. doi:10.1002/adsc.200900671 |
| 7. | Argüello, J. E.; Schmidt, L. C.; Peñéñory, A. B. Org. Lett. 2003, 5, 4133–4136. doi:10.1021/ol035545n |
| 8. | Argüello, J. E.; Schmidt, L. C.; Peñéñory, A. B. J. Org. Chem. 2007, 72, 2936–2944. doi:10.1021/jo062569+ |
| 35. | Tao, C.; Lv, A.; Zhao, N.; Yang, S.; Liu, X.; Zhou, J.; Liu, W.; Zhao, J. Synlett 2011, 134–138. doi:10.1055/s-0030-1259079 |
| 4. | Rossi, R. A.; Pierini, A. B.; Peñéñory, A. B. Chem. Rev. 2003, 103, 71–168. doi:10.1021/cr960134o |
| 5. | Peñéñory, A. B.; Argüello, J. E. In Handbook of Synthetic Photochemistry; Albini, A.; Fagnoni, M., Eds.; Wiley-VCH Verlag GmbH &Co: Weinheim, 2010; pp 319–346. |
| 31. | Ke, F.; Qu, Y.; Jiang, Z.; Li, Z.; Wu, D.; Zhou, X. Org. Lett. 2011, 13, 454–457. doi:10.1021/ol102784c |
| 32. | Reddy, K. H. V.; Reddy, V. P.; Kumar, A. A.; Kranthi, G.; Nageswar, Y. V. D. Beilstein J. Org. Chem. 2011, 7, 886–891. doi:10.3762/bjoc.7.101 |
| 17. | Yoon, H.-J.; Choi, J.-W.; Kang, H.; Kang, T.; Lee, S.-M.; Jun, B.-H.; Lee, Y.-S. Synlett 2010, 2518–2522. doi:10.1055/s-0030-1258545 |
| 18. | Jammi, S.; Barua, P.; Rout, L.; Saha, P.; Punniyamurthy, T. Tetrahedron Lett. 2008, 49, 1484–1487. doi:10.1016/j.tetlet.2007.12.118 |
| 21. | Wang, L.; Zhou, W.-Y.; Chen, S.-C.; He, M.-Y.; Chen, Q. Adv. Synth. Catal. 2012, 354, 839–845. doi:10.1002/adsc.201100788 |
| 22. | Fu, C.-F.; Liu, Y.-H.; Peng, S.-M.; Liu, S.-T. Tetrahedron 2010, 66, 2119–2122. doi:10.1016/j.tet.2010.01.081 |
| 23. | Fernández-Rodríguez, M. A.; Hartwig, J. F. J. Org. Chem. 2009, 74, 1663–1672. doi:10.1021/jo802594d |
| 58. | Zheng, J.; Liu, X.; Quan, Q.; Shin, Y.-J.; Sun, D.; Lu, Y. Org. Biomol. Chem. 2010, 1293–1295. doi:10.1039/b926217b |
| 13. | Ley, S. V.; Thomas, A. W. Angew. Chem., Int. Ed. 2003, 42, 5400–5449. doi:10.1002/anie.200300594 |
| 14. | Beletskaya, I. P.; Cheprakov, A. V. Coord. Chem. Rev. 2004, 248, 2337–2364. doi:10.1016/j.ccr.2004.09.014 |
| 15. | Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2008, 47, 3096–3099. doi:10.1002/anie.200703209 |
| 16. | Qiao, J. X.; Lam, P. Y. S. Synthesis 2011, 829–856. doi:10.1055/s-0030-1258379 |
| 24. | Kwong, F. Y.; Buchwald, S. L. Org. Lett. 2002, 4, 3517–3520. doi:10.1021/ol0266673 |
| 25. | Bates, C. G.; Gujadhur, R. K.; Venkataraman, D. Org. Lett. 2002, 4, 2803–2806. doi:10.1021/ol0264105 |
| 26. | Sperotto, E.; van Klink, G. P. M.; de Vries, J. G.; van Koten, G. J. Org. Chem. 2008, 73, 5625–5628. doi:10.1021/jo800491k |
| 27. | Herrero, M. T.; SanMartín, R.; Domínguez, E. Tetrahedron 2009, 65, 1500–1503. doi:10.1016/j.tet.2008.11.062 |
| 28. | Jammi, S.; Sakthivel, S.; Rout, L.; Mukherjee, T.; Mandal, S.; Mitra, R.; Saha, P.; Punniyamurthy, T. J. Org. Chem. 2009, 74, 1971–1976. doi:10.1021/jo8024253 |
| 29. | Feng, Y.-S.; Qi, H.-X.; Wang, W.-C.; Liang, Y.-F.; Xu, H.-J. Tetrahedron Lett. 2012, 53, 2914–2917. doi:10.1016/j.tetlet.2012.04.004 |
| 52. | Sawada, N.; Itoh, T.; Yasuda, N. Tetrahedron Lett. 2006, 47, 6595–6597. doi:10.1016/j.tetlet.2006.07.008 |
| 12. | Hartwig, J. F. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E.-I., Ed.; Wiley-Interscience: New York, 2002; Vol. 1, pp 1051–1096. |
| 52. | Sawada, N.; Itoh, T.; Yasuda, N. Tetrahedron Lett. 2006, 47, 6595–6597. doi:10.1016/j.tetlet.2006.07.008 |
| 10. | Kondo, T.; Mitsudo, T.-a. Chem. Rev. 2000, 100, 3205–3220. doi:10.1021/cr9902749 |
| 11. | Beletskaya, I. P.; Ananikov, V. P. Chem. Rev. 2011, 111, 1596–1636. doi:10.1021/cr100347k |
| 19. | Akkilagunta, V. K.; Reddy, V. P.; Rao, K. R. Synlett 2010, 1260–1264. doi:10.1055/s-0029-1219801 |
| 20. | Correa, A.; Carril, M.; Bolm, C. Angew. Chem. 2008, 120, 2922–2925. doi:10.1002/ange.200705668 |
| 57. | Iyer, R. P.; Egan, W.; Regan, J. B.; Beaucage, S. L. J. Am. Chem. Soc. 1990, 112, 1253–1254. doi:10.1021/ja00159a059 |
| 40. | Xu, H.-J.; Liang, Y.-F.; Cai, Z.-Y.; Qi, H.-X.; Yang, C.-Y.; Feng, Y.-S. J. Org. Chem. 2011, 76, 2296–2300. doi:10.1021/jo102506x |
| 41. | Bhakuni, B. S.; Balkrishna, S. J.; Kumar, A.; Kumar, S. Tetrahedron Lett. 2012, 53, 1354–1357. doi:10.1016/j.tetlet.2012.01.003 |
| 36. | Akkilagunta, V. K.; Kakulapati, R. R. J. Org. Chem. 2011, 76, 6819–6824. doi:10.1021/jo200793k |
| 37. | Prasad, D. J. C.; Sekar, G. Org. Lett. 2011, 13, 1008–1011. doi:10.1021/ol103041s |
| 38. | Ma, D.; Xie, S.; Xue, P.; Zhang, X.; Dong, J.; Jiang, Y. Angew. Chem., Int. Ed. 2009, 48, 4222–4225. doi:10.1002/anie.200900486 |
| 39. | Li, Y.; Nie, C.; Wang, H.; Li, X.; Verpoort, F.; Duan, C. Eur. J. Org. Chem. 2011, 7331–7338. doi:10.1002/ejoc.201101121 |
| 52. | Sawada, N.; Itoh, T.; Yasuda, N. Tetrahedron Lett. 2006, 47, 6595–6597. doi:10.1016/j.tetlet.2006.07.008 |
| 52. | Sawada, N.; Itoh, T.; Yasuda, N. Tetrahedron Lett. 2006, 47, 6595–6597. doi:10.1016/j.tetlet.2006.07.008 |
| 50. | Petrillo, G.; Novi, M.; Garbarino, G.; Filiberti, M. Tetrahedron 1989, 45, 7411–7420. doi:10.1016/S0040-4020(01)89203-6 |
| 51. | Lai, C.; Backes, B. J. Tetrahedron Lett. 2007, 48, 3033–3037. doi:10.1016/j.tetlet.2007.02.128 |
| 48. | Nishiguchi, A.; Maeda, K.; Miki, S. Synthesis 2006, 4131–4134. doi:10.1055/s-2006-950353 |
| 49. | Ho, D. K. H.; Chan, L.; Hooper, A.; Brennan, P. E. Tetrahedron Lett. 2011, 52, 820–823. doi:10.1016/j.tetlet.2010.12.050 |
| 42. | Evindar, G.; Batey, R. A. J. Org. Chem. 2006, 71, 1802–1808. doi:10.1021/jo051927q |
| 43. | Ma, H. C.; Jiang, X. Z. Synlett 2008, 1335–1340. doi:10.1055/s-2008-1072764 |
| 44. | Murru, S.; Ghosh, H.; Sahoo, S. K.; Patel, B. K. Org. Lett. 2009, 11, 4254–4257. doi:10.1021/ol9017535 |
| 45. | Ma, D.; Geng, Q.; Zhang, H.; Jiang, Y. Angew. Chem., Int. Ed. 2010, 49, 1291–1294. doi:10.1002/anie.200905646 |
| 46. | Ma, D.; Lu, X.; Shi, L.; Zhang, H.; Jiang, Y.; Liu, X. Angew. Chem., Int. Ed. 2011, 50, 1118–1121. doi:10.1002/anie.201005787 |
| 47. | Li, J.; Zhang, Y.; Jiang, Y.; Ma, D. Tetrahedron Lett. 2012, 53, 2511–2513. doi:10.1016/j.tetlet.2012.03.006 |
© 2013 Soria-Castro and Peñéñory; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)