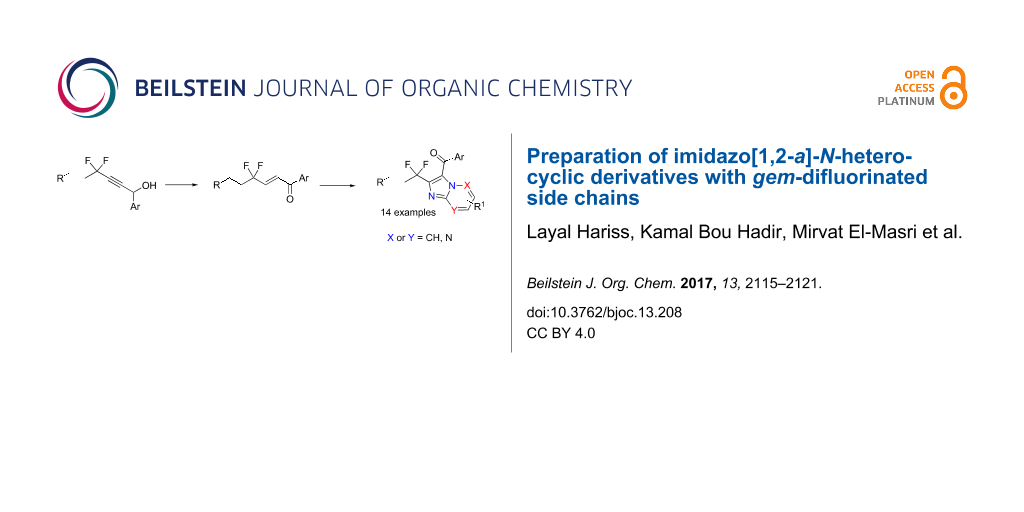
Abstract
Using an aerobic oxidative coupling, different new imidazo[1,2-a]-N-heterocycles with gem-difluroroalkyl side chains have been prepared in fair yields by the reaction of gem-difluoroenones with aminopyridines, -pyrimidines and -pyridazines. Condensed heterocycles of this type play an important role as key core structures of various bioactive compounds. Further, starting with a chloroimidazopyridazine derivative, Pd-catalyzed coupling reactions as well as nucleophilic substitutions have been performed successfully in order to increase the molecular diversity.

Graphical Abstract
Introduction
Nitrogen-containing heterocyclic compounds are frequently found in bioactive naturally occurring compounds, as well as in the synthetic pharmacopeia. Imidazo[1,2-a]pyridine is an important heterocyclic system present in many molecules featuring diverse biological activities, such as antiviral, antimicrobial, antitumor, anti-inflammatory, antiparasitic, hypnotic, etc. [1-5]. It is recognized as a key scaffold due to its broad occurrence in a number of drug candidates and drugs, such as zolpidem [6] (1a, used in the treatment of insomnia), and alpidem [6] (1b, an anxiolytic agent). Some imidazopyridine derivatives also act as β-amyloid formation inhibitors, GABA and benzodiazepine receptor agonists, and cardiotonic agents [7-10]. Further, the biological activities of imidazo[1,2-a]pyridines proved to be strongly depending upon the nature of substituents at C2 and C3 positions. For instance, the 3-aroylimidazo[1,2-a]pyridines 1c demonstrated also good anticancer properties [11,12], while imidazo[1,2-a]pyrimidines 2 are also known for their antituberculosis activity [13], and imidazopyridazine 3 acts as a sirtuin modulator [14].
On the other hand, the incorporation of fluorine or fluorinated groups into organic molecules has been widely recognized as a general strategy toward drug development in pharmaceutical research. This is connected to fluorine's electronegativity, size, and lipophilicity [15,16], which can strongly improve the biological properties of molecules through, for instance, increase of metabolic stability and bioavailability for many drugs and pharmacological tools. So, the preparation of fluorinated molecules is a very attractive research area for organic and medicinal chemists [17-20].
Our research program aims to synthesize new fluorinated molecules based on the easy access and the versatility of fluorinated propargylic derivatives [21]. Thus, taking into account the known biological properties of the imidazo-fused N-heterocycles, we became interested in the preparation of new derivatives of this type possessing gem-difluorinated side chains as indicated in Figure 1. Such new fluorinated heterocycles could be of interest for bioorganic and medicinal chemistry studies.
![[1860-5397-13-208-1]](/bjoc/content/figures/1860-5397-13-208-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative examples of bioactive imidazo[1,2-a]pyridines, imidazo[1,2-a]pyrimidines, imidazopyridazines and our target molecules.
Figure 1: Representative examples of bioactive imidazo[1,2-a]pyridines, imidazo[1,2-a]pyrimidines, imidazopyr...
Several synthetic approaches for imidazopyridines are available, but only a few examples have been reported to date for the construction of this scaffold with introduction of fluorine [22], trifluoromethyl [23] or trifluoroethyl groups [24]. Herein, we report the synthesis of imidazo[1,2-a]pyridines, imidazo[1,2-a]pyrimidines, and imidazopyridazines with fluorinated side chains following an efficient strategy developed by Hajra et al. [25]. This methodology, developed for the synthesis of 3-aroylimidazopyridines, involves a copper(II) acetate-catalyzed aerobic oxidative amination and it proceeds through a tandem Michael addition followed by an intramolecular oxidative amination. Therefore, our target molecules A could be synthesized by the oxidative coupling of 2-aminopyridines with α,β-unsaturated ketones B, themselves easily accessible from gem-difluoropropargylic alcohols C through a base-mediated isomerization process (Scheme 1) [26,27].
![[1860-5397-13-208-i1]](/bjoc/content/inline/1860-5397-13-208-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic scheme for the preparation of our target molecules A.
Scheme 1: Retrosynthetic scheme for the preparation of our target molecules A.
Results and Discussion
The required propargylic alcohols 5a–e (type C, Scheme 1) were obtained in 27–73% yields by reaction of the lithium salt of the easily accessible gem-difluoro propargylic derivatives 4 [28] with aromatic aldehydes. Then, the DBU-mediated isomerization afforded the desired enones 6a–e in 21–66% yields (Scheme 2 and Table 1).
![[1860-5397-13-208-i2]](/bjoc/content/inline/1860-5397-13-208-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of enones 6 with a gem-difluoroalkyl side chain.
Scheme 2: Synthesis of enones 6 with a gem-difluoroalkyl side chain.
For the synthesis of the desired nitrogen heterocycles, we started our study by reacting 6a with 2-aminopyridine in the presence of AlCl3 and I2 under an O2 atmosphere [29]. However, only a poor yield was obtained (20%, Scheme 3).
![[1860-5397-13-208-i3]](/bjoc/content/inline/1860-5397-13-208-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Then, we found that Cu(OAc)2·H2O (10 mol %) and 1,10-phenanthroline (10 mol %) in chlorobenzene at 160 °C under an O2 atmosphere, following the conditions recently reported by Hajra et al. [25], gave 7a in 62% yield (Table 2, entry 1). Having these optimized conditions in hand, and to explore the substrate scope, different substituted 2-aminopyridines were successfully employed to afford the tandem oxidative cyclization products 7 in 32–65% yields. On the other hand, enones 6 with two different R groups (Table 2, entries 1, 2, 3, 6, 8, and 9) were well tolerated under the optimized conditions affording the tandem products 7 in fair to moderate yields, although lower yields were obtained in the cases of 7j and 7g (32% and 36%, respectively). Moreover, two other important heterocyclic frameworks, imidazo[1,2-a]pyrimidines 7d and imidazo[1,2-b]pyridazines 7e, have been synthesized by the same method albeit in slightly decreased yields (Table 2, entries 4 and 5).
Table 2: Preparation of different imidazo[1,2-a]-N-heterocyclic derivatives.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-208-i5.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | Product | Ar | R | R1 | X | Y | Time | Yield (%) |
| 1 | 7a | Ph | Ph | H | CH | CH | 25 h | 62 |
| 2 | 7b | Ph | Ph | 7-Me | CH | CH | 20 h | 60 |
| 3 | 7c | Ph | Ph | 6-Br | CH | CH | 46 h | 60 |
| 4 | 7d | Ph | Ph | H | CH | N | 30 h | 57 |
| 5 | 7e | Ph | Ph | 6-Cl | N | CH | 24 h | 53 |
| 6 | 7f | Ph | -CH2OBn | H | CH | CH | 33 h | 55 |
| 7 | 7g | Ph | -CH2OBn | H | CH | N | 29 h | 36 |
| 8 | 7h | o-BrPh | Ph | H | CH | CH | 4 h | 65 |
| 9 | 7i | 2-naphthaldehyde | Ph | H | CH | CH | 3.5 h | 59 |
| 10 | 7j | p-MeOPh | Ph | H | CH | CH | 6 h | 32 |
The structures of molecules 7 are in full agreement with their spectroscopical (NMR) and analytical data (HRMS). For the imidazopyridines, the structure of 7a was confirmed by X-ray analysis (Figure 2) [30] and the other derivatives were proposed by analogy. In the same way as for the imidazopyridazines, the structure of 7e was established by X-ray analysis [30] and this was extended to the other derivatives. These results unambiguously demonstrate the regiochemistry of the reaction. These cascade reactions proceed first through a Michael addition of the primary amine on the enone, followed by an intramolecular cyclization by the pyridine/pyrimidine nucleus. Unfortunately, no crystal structure could be obtained for the imidazopyrimidines and therefore the corresponding structures 7d and 7g were proposed by analogy.
![[1860-5397-13-208-2]](/bjoc/content/figures/1860-5397-13-208-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of 7a and 7e by X-ray crystallography analysis.
Figure 2: Structures of 7a and 7e by X-ray crystallography analysis.
These oxidative coupling conditions appeared compatible with the first isomerization step, therefore, the possibility of a "one-pot" reaction was considered. Indeed, by heating alcohol 5a (Table 1, entry 1) with 2-aminopyridine and DBU (1,8-diazabicycloundec-7-ene) under the same conditions as mentioned above, the desired imidazopyridine derivative 7a was isolated in 33% yield (Scheme 4). This one-pot process gives an overall yield very close to the two-step reaction (38%).
![[1860-5397-13-208-i4]](/bjoc/content/inline/1860-5397-13-208-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Further, the halogen-substituted substrate 7e appeared as an attractive precursor to increase the molecular diversity around this scaffold. In order to explore this possibility, we performed two Suzuki–Miyaura reactions, as representative examples of Pd-catalyzed coupling processes (Table 3). They gave the target molecules 8 and 9 in 46% and 53% yields, respectively. On the other hand, two nucleophilic substitution reactions using phenol and morpholine gave the expected heterocycles 10 and 11 in 73% and 23% yields, respectively.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-208-i6.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-208-i7.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-208-i8.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-208-i9.svg?max-width=637&scale=1.0)
Conclusion
In summary, we developed a short and completely regioselective method for the synthesis of imidazo[1,2-a]-N-heterocycles with gem-fluorinated side chains starting from easily accessible propargylic fluorides. Although the yields are only moderate to fair, this short (1–2 steps) method offers significant flexibility to prepare focused libraries of molecules with this core structure. Such new fluorine-containing heteroaromatic frameworks would be of much interest for biological studies in different areas of life sciences.
Experimental
Representative procedure for the synthesis of imidazopyridine 7a
The syntheses of propargylic fluorides 5 and enones 6 were performed in a similar way as described before [26].
Synthesis of 4,4-difluoro-1,6-diphenylhex-2-yn-1-ol (5a)
To a solution of gem-difluoro intermediate 4 [28] (500 mg, 2.77 mmol, 1 equiv) in anhydrous THF (6 mL) cooled at −80 °C was added dropwise under nitrogen a 2.5 M solution of n-BuLi in hexanes (1.3 mL, 3.30 mmol, 1.2 equiv). The mixture was stirred for 1 h at a temperature below −80 °C before dropwise addition of the aldehyde (0.35 mL, 3.33 mmol, 1.2 equiv) in anhydrous THF (4 mL). The reaction mixture was stirred for additional 45 min at t < −80 °C and then allowed to warm to rt for 2 h. The mixture was then treated with a saturated ammonium chloride solution and extracted with ethyl acetate. The combined organic phases were washed with water, dried over Na2SO4 and concentrated in vacuo. The crude product was purified by chromatography on silica gel, using a mixture of petroleum ether/ethyl acetate as eluent. After purification by chromatography on silica gel, propargylic alcohol 5a was obtained as a colorless oil (580 g, 73% yield); Rf 0.46 (petroleum ether/AcOEt 8:2); 1H NMR (CDCl3, 300 MHz) δ 7.59–7.25 (m, 10H), 5.54 (t, JHF = 3.9 Hz, 1H), 2.99–2.93 (m, 2H), 2.54–2.38 (m, 2H), 2.83 (br s, 1H); 13C NMR (CDCl3, 75 MHz) δ 139.7, 138.9 (t, 4J = 1.1 Hz), 128.8, 128.7 (2C), 128.5 (2C), 128.3 (2C), 126.5, 126.3, 125.9, 114.1 (t, 1J = 233.4 Hz), 87.0 (t, 3J = 6.8 Hz), 79.1 (t, 2J = 40.9 Hz), 64.0 (t, 4J = 1.8 Hz), 40.8 (t, 2J = 26.1 Hz), 28.9 (t, 3J = 4.0 Hz); 19F NMR (CDCl3, 282 MHz) δ −83.45 (td, JFH = 14.6, 3.9 Hz); HRMS (ESI) m/z [M + Na]+: calcd. for C18H16OF2Na, 309.10614; found, 309.1060 (0 ppm); m/z [M – HF + Na]+: calcd. for C18H15OFNa, 289.09991; found, 289.0992 (2 ppm).
Synthesis of (E)-4,4-difluoro-1,6-diphenylhex-2-en-1-one (6a)
The previous difluoropropargylic alcohol 5a (540 mg, 1.88 mmol, 1 equiv) was dissolved in THF (4 mL), then DBU (0.42 mL, 2.82 mmol, 1.5 equiv) was added and the reaction mixture was stirred at room temperature. After 2 h, 19F NMR showed 100% conversion and the reaction mixture was neutralized with a saturated solution of NH4Cl. After extraction with ethyl acetate, the organic phases were washed with water, dried (Na2SO4) and concentrated in vacuo. The crude product was purified by chromatography on silica gel, using a mixture of petroleum ether/ethyl acetate as eluent. Enone 6a was isolated as a colorless oil (335 mg, 62% yield); Rf 0.43 (petroleum ether/AcOEt 9:1); 1H NMR (CDCl3, 500 MHz) δ 8.04 (m, 2H), 7.66–7.64 (m, 1H), 7.57–7.52 (m, 2H), 7.35–7.28 (m, 6H), 6.97 (m, 1H), 2.93–2.92 (m, 2H), 2.41–2.35 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 188.93, 139.8, 138.3 (t, 2J = 27.1 Hz), 136.7, 133.5, 128.7 (2C), 128.6 (2C), 128.5 (2C), 128.1 (2C), 127.6 (t, 3J = 7.5 Hz), 126.3, 120.6 (t, 1J = 240.4 Hz), 38.9 (t, 2J = 25.9 Hz), 28.2 (t, 3J = 4.3 Hz); 19F NMR (CDCl3, 282 MHz) δ −98.84 (m); HRMS (ESI) m/z [M + Na]+: calcd. for C18H16OF2Na, 309.10614; found, 309.1059 (1 ppm).
Synthesis of (2-(1,1-difluoro-3-phenylpropyl)imidazo[1,2-a]pyridin-3-yl)(phenyl)methanone (7a)
A mixture of 2-aminopyridine (20 mg, 0.21 mmol, 1.2 equiv), enone 6a (51 mg, 0.17 mmol, 1 equiv), Cu(OAc)2·H2O (3.6 mg, 0.02 mmol, 10 mol %), and 1,10-phenanthroline (2.5 μL, 0.02 mmol, 10 mol %) in chlorobenzene (1 mL) was stirred in a reaction tube at 160 °C under an O2 atmosphere. After 25 h, 19F NMR monitoring indicated complete consumption of the starting material. The reaction mixture was cooled to room temperature, filtered and extracted with dichloromethane. The filtrate was concentrated and the crude product was purified by column chromatography on silica gel, using petroleum ether/ethyl acetate as eluent. 7a was isolated as white crystals (32 mg, 62% yield); Rf 0.46 (petroleum ether/EtOAc 7:3); Mp: 117 °C; 1H NMR (CDCl3, 300 MHz) δ 8.76 (d, J = 6.9 Hz, 1H), 7.89 (s, 1H), 7.86 (s, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.64 (t, J = 7.3 Hz, 1H), 7.53–7.43 (m, 3H), 7.26–7.13 (m, 5H), 7.02 (t, J = 6.7 Hz, 1H), 2.72–2.65 (m, 4H); 13C NMR (CDCl3, 75 MHz) δ 187.9, 146.1, 145.1, 140.4, 139.5 (t, 3J = 2.1 Hz), 133.3 (2C), 129.5, 128.4 (2C), 128.3 (3C), 128.2 (2C), 127.3, 126.0, 120.6, 120.3 (t, 1J = 239.8 Hz), 118.2, 114.7, 39.3 (t, 2J = 25.1 Hz), 28.3 (t, 3J = 4.4 Hz); 19F NMR (CDCl3, 282 MHz) δ −90.93 (t, J = 15.7 Hz); HRMS (ESI) m/z [M + Na]+: calcd. for C23H18N2OF2Na, 399.12794; found, 399.1279 (0 ppm); m/z [M + H]+: calcd. for C23H19N2OF2, 377.14599; found, 377.1454 (2 ppm); m/z [M – HF + Na]+: calcd. for C23H17N2OFNa, 379.12171; found, 379.1216 (0 ppm).
One pot synthesis of (2-(1,1-difluoro-3-phenylpropyl)imidazo[1,2-a]pyridin-3-yl)(phenyl)methanone (7a)
A mixture of 2-aminopyridine (25 mg, 0.26 mmol, 2.5 equiv), alcohol 5a (30 mg, 0.10 mmol, 1 equiv), DBU (0.03 mL, 0.20 mmol, 2 equiv), Cu(OAc)2·H2O (2.1 mg, 0.01 mmol, 10 mol %), and 1,10-phenanthroline (1.4 μL, 0.01 mmol, 10 mol %) in chlorobenzene (1 mL) was stirred in a reaction tube at 160 °C under an O2 atmosphere. After 4 h monitoring by 19F NMR indicated the disappearance of the starting material. Thus the mixture was cooled to room temperature, filtered, washed and extracted with dichloromethane. The organic phase was concentrated and the crude product was purified by column chromatography on silica gel, using petroleum ether/ethyl acetate as eluent. Imidazopyridine 7a was isolated in 33% yield.
Supporting Information
| Supporting Information File 1: Experimental details and characterization data of new compounds with copies of 1H, 13C and 19F NMR spectra. | ||
| Format: PDF | Size: 6.6 MB | Download |
References
-
Lhassani, M.; Chavignon, O.; Chezal, J.-M.; Teulade, J.-C.; Chapat, J.-P.; Snoeck, R.; Andrei, G.; Balzarini, J.; De Clercq, E.; Gueiffier, A. Eur. J. Med. Chem. 1999, 34, 271. doi:10.1016/S0223-5234(99)80061-0
and references cited therein.
Return to citation in text: [1] -
Pericherla, K.; Kaswan, P.; Pandey, K.; Kumar, A. Synthesis 2015, 47, 887. doi:10.1055/s-0034-1380182
and references cited therein.
Return to citation in text: [1] -
Koubachi, J.; El Kazzouli, S.; Bousmina, M.; Guillaumet, G. Eur. J. Org. Chem. 2014, 5119. doi:10.1002/ejoc.201400065
and references cited therein.
Return to citation in text: [1] -
Basilio-Lopes, A.; de Aquino, T. M.; Mongeot, A.; Bourguignon, J.-J.; Schmitt, M. Tetrahedron Lett. 2012, 53, 2583. doi:10.1016/j.tetlet.2012.02.117
and references cited therein.
Return to citation in text: [1] -
Rao, N. S.; Kistareddy, C.; Balram, B.; Ram, B. Pharma Chem. 2012, 4, 2408.
and references cited therein.
Return to citation in text: [1] -
Langer, S. Z.; Arbilla, S.; Benavides, J.; Scatton, B. Adv. Biochem. Psychopharmacol. 1990, 46, 61.
and references cited therein.
Return to citation in text: [1] [2] -
Humphries, A. C.; Gancia, E.; Gilligan, M. T.; Goodacre, S.; Hallett, D.; Marchant, K. J.; Thomas, S. R. Bioorg. Med. Chem. Lett. 2006, 16, 1518. doi:10.1016/j.bmcl.2005.12.037
Return to citation in text: [1] -
Fuchs, K.; Romig, M.; Mendla, K.; Briem, H.; Fechteler, K. Novel beta-amyloid inhibitors, method for producing the same and the use thereof as medicaments. WO Patent WO2002014313, July 21, 2001.
Return to citation in text: [1] -
Davey, D.; Erhardt, P. W.; Lumma, W. C., Jr.; Wiggins, J.; Sullivan, M.; Pang, D.; Cantor, E. J. Med. Chem. 1987, 30, 1337. doi:10.1021/jm00391a012
Return to citation in text: [1] -
Fookes, C. J. R.; Pham, T. Q.; Mattner, F.; Greguric, I.; Loch, C.; Liu, X.; Berghofer, P.; Shepherd, R.; Gregoire, M.-C.; Katsifis, A. J. Med. Chem. 2008, 51, 3700. doi:10.1021/jm7014556
Return to citation in text: [1] -
Tung, Y.-S.; Coumar, M. S.; Wu, Y.-S.; Shiao, H.-Y.; Chang, J.-Y.; Liou, J.-P.; Shukla, P.; Chang, C.-W.; Chang, C.-Y.; Kuo, C.-C.; Yeh, T.-K.; Lin, C.-Y.; Wu, J.-S.; Wu, S.-Y.; Liao, C.-C.; Hsieh, H.-P. J. Med. Chem. 2011, 54, 3076. doi:10.1021/jm101027s
Return to citation in text: [1] -
Hseih, H.-P.; Chao, Y.-S.; Liou, J.-P.; Chang, J.-Y.; Tung, Y.-S. Antitumor compounds. U.S. Patent US7,456,289, Dec 15, 2005.
Return to citation in text: [1] -
Moraski, G. C.; Markley, L. D.; Hipskind, P. A.; Boshoff, H.; Cho, S.; Franzblau, S. G.; Miller, M. J. ACS Med. Chem. Lett. 2011, 2, 466. doi:10.1021/ml200036r
Return to citation in text: [1] -
Casaubon, R. L.; Narayan, R.; Oalmann, C.; Vu, C. B. Substituted bicyclic azaheterocycles and analogues as sirtuin modulators. WO Patent WO2,013,059,587, Oct 19, 2012.
Return to citation in text: [1] -
Hudlicky, M., Ed. Chemistry of Organic Fluorine Compounds II: A Critical Review; ACS Monograph, Vol. 187; American Chemical Society: Washington, DC, 1995.
Return to citation in text: [1] -
Kitazume, T.; Yamazaki, T. Experimental Methods in Organic Fluorine Chemistry; Gordon and Breach Science Publishers: Tokyo, 1998.
Return to citation in text: [1] -
Qing, F.-L.; Zheng, F. Synlett 2011, 1052. doi:10.1055/s-0030-1259947
and references cited therein. See for a recent review article.
Return to citation in text: [1] -
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315. doi:10.1021/acs.jmedchem.5b00258
and references cited therein. See for a recent review article.
Return to citation in text: [1] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422. doi:10.1021/acs.chemrev.5b00392
and references cited therein. See for a recent review article.
Return to citation in text: [1] -
Yerien, D. E.; Bonesi, S.; Postigo, A. Org. Biomol. Chem. 2016, 14, 8398. doi:10.1039/c6ob00764c
and references cited therein. See for a recent review article.
Return to citation in text: [1] -
Hachem, A.; Grée, D.; Chandrasekhar, S.; Grée, R. Synthesis 2017, 49, 2101. doi:10.1055/s-0036-1589484
and references cited therein.
Return to citation in text: [1] -
Liu, P.; Gao, Y.; Gu, W.; Shen, Z.; Sun, P. J. Org. Chem. 2015, 80, 11559. doi:10.1021/acs.joc.5b01961
Return to citation in text: [1] -
Monir, K.; Bagdi, A. K.; Ghosh, M.; Hajra, A. J. Org. Chem. 2015, 80, 1332. doi:10.1021/jo502928e
Return to citation in text: [1] -
Zhu, M.; Han, X.; Fu, W.; Wang, Z.; Ji, B.; Hao, X.-Q.; Song, M.-P.; Xu, C. J. Org. Chem. 2016, 81, 7282. doi:10.1021/acs.joc.6b00950
Return to citation in text: [1] -
Monir, K.; Bagdi, A. K.; Mishra, S.; Majee, A.; Hajra, A. Adv. Synth. Catal. 2014, 356, 1105. doi:10.1002/adsc.201300900
Return to citation in text: [1] [2] -
Nasr El Dine, A.; Khalaf, A.; Grée, D.; Tasseau, O.; Fares, F.; Jaber, N.; Lesot, P.; Hachem, A.; Grée, R. Beilstein J. Org. Chem. 2013, 9, 1943. doi:10.3762/bjoc.9.230
Return to citation in text: [1] [2] -
Nasr El Dine, A.; Grée, D.; Roisnel, T.; Caytan, E.; Hachem, A.; Grée, R. Eur. J. Org. Chem. 2016, 556. doi:10.1002/ejoc.201501347
Return to citation in text: [1] -
Pujari, S. A.; Kaliappan, K. P.; Valleix, A.; Grée, D.; Grée, R. Synlett 2008, 2503. doi:10.1055/s-2008-1078179
Return to citation in text: [1] [2] -
Xing, M.-M.; Xin, M.; Shen, C.; Gao, J.-R.; Jia, J.-H.; Li, Y.-J. Tetrahedron 2016, 72, 4201. doi:10.1016/j.tet.2016.05.052
Return to citation in text: [1] -
CCDC 1549632 (for compound 7a) and CCDC 1549633 (for compound 7e) contain the supplementary crystallographical data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre.
Return to citation in text: [1] [2]
| 30. | CCDC 1549632 (for compound 7a) and CCDC 1549633 (for compound 7e) contain the supplementary crystallographical data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre. |
| 29. | Xing, M.-M.; Xin, M.; Shen, C.; Gao, J.-R.; Jia, J.-H.; Li, Y.-J. Tetrahedron 2016, 72, 4201. doi:10.1016/j.tet.2016.05.052 |
| 25. | Monir, K.; Bagdi, A. K.; Mishra, S.; Majee, A.; Hajra, A. Adv. Synth. Catal. 2014, 356, 1105. doi:10.1002/adsc.201300900 |
| 1. |
Lhassani, M.; Chavignon, O.; Chezal, J.-M.; Teulade, J.-C.; Chapat, J.-P.; Snoeck, R.; Andrei, G.; Balzarini, J.; De Clercq, E.; Gueiffier, A. Eur. J. Med. Chem. 1999, 34, 271. doi:10.1016/S0223-5234(99)80061-0
and references cited therein. |
| 2. |
Pericherla, K.; Kaswan, P.; Pandey, K.; Kumar, A. Synthesis 2015, 47, 887. doi:10.1055/s-0034-1380182
and references cited therein. |
| 3. |
Koubachi, J.; El Kazzouli, S.; Bousmina, M.; Guillaumet, G. Eur. J. Org. Chem. 2014, 5119. doi:10.1002/ejoc.201400065
and references cited therein. |
| 4. |
Basilio-Lopes, A.; de Aquino, T. M.; Mongeot, A.; Bourguignon, J.-J.; Schmitt, M. Tetrahedron Lett. 2012, 53, 2583. doi:10.1016/j.tetlet.2012.02.117
and references cited therein. |
| 5. |
Rao, N. S.; Kistareddy, C.; Balram, B.; Ram, B. Pharma Chem. 2012, 4, 2408.
and references cited therein. |
| 11. | Tung, Y.-S.; Coumar, M. S.; Wu, Y.-S.; Shiao, H.-Y.; Chang, J.-Y.; Liou, J.-P.; Shukla, P.; Chang, C.-W.; Chang, C.-Y.; Kuo, C.-C.; Yeh, T.-K.; Lin, C.-Y.; Wu, J.-S.; Wu, S.-Y.; Liao, C.-C.; Hsieh, H.-P. J. Med. Chem. 2011, 54, 3076. doi:10.1021/jm101027s |
| 12. | Hseih, H.-P.; Chao, Y.-S.; Liou, J.-P.; Chang, J.-Y.; Tung, Y.-S. Antitumor compounds. U.S. Patent US7,456,289, Dec 15, 2005. |
| 26. | Nasr El Dine, A.; Khalaf, A.; Grée, D.; Tasseau, O.; Fares, F.; Jaber, N.; Lesot, P.; Hachem, A.; Grée, R. Beilstein J. Org. Chem. 2013, 9, 1943. doi:10.3762/bjoc.9.230 |
| 27. | Nasr El Dine, A.; Grée, D.; Roisnel, T.; Caytan, E.; Hachem, A.; Grée, R. Eur. J. Org. Chem. 2016, 556. doi:10.1002/ejoc.201501347 |
| 7. | Humphries, A. C.; Gancia, E.; Gilligan, M. T.; Goodacre, S.; Hallett, D.; Marchant, K. J.; Thomas, S. R. Bioorg. Med. Chem. Lett. 2006, 16, 1518. doi:10.1016/j.bmcl.2005.12.037 |
| 8. | Fuchs, K.; Romig, M.; Mendla, K.; Briem, H.; Fechteler, K. Novel beta-amyloid inhibitors, method for producing the same and the use thereof as medicaments. WO Patent WO2002014313, July 21, 2001. |
| 9. | Davey, D.; Erhardt, P. W.; Lumma, W. C., Jr.; Wiggins, J.; Sullivan, M.; Pang, D.; Cantor, E. J. Med. Chem. 1987, 30, 1337. doi:10.1021/jm00391a012 |
| 10. | Fookes, C. J. R.; Pham, T. Q.; Mattner, F.; Greguric, I.; Loch, C.; Liu, X.; Berghofer, P.; Shepherd, R.; Gregoire, M.-C.; Katsifis, A. J. Med. Chem. 2008, 51, 3700. doi:10.1021/jm7014556 |
| 28. | Pujari, S. A.; Kaliappan, K. P.; Valleix, A.; Grée, D.; Grée, R. Synlett 2008, 2503. doi:10.1055/s-2008-1078179 |
| 6. |
Langer, S. Z.; Arbilla, S.; Benavides, J.; Scatton, B. Adv. Biochem. Psychopharmacol. 1990, 46, 61.
and references cited therein. |
| 24. | Zhu, M.; Han, X.; Fu, W.; Wang, Z.; Ji, B.; Hao, X.-Q.; Song, M.-P.; Xu, C. J. Org. Chem. 2016, 81, 7282. doi:10.1021/acs.joc.6b00950 |
| 6. |
Langer, S. Z.; Arbilla, S.; Benavides, J.; Scatton, B. Adv. Biochem. Psychopharmacol. 1990, 46, 61.
and references cited therein. |
| 25. | Monir, K.; Bagdi, A. K.; Mishra, S.; Majee, A.; Hajra, A. Adv. Synth. Catal. 2014, 356, 1105. doi:10.1002/adsc.201300900 |
| 17. |
Qing, F.-L.; Zheng, F. Synlett 2011, 1052. doi:10.1055/s-0030-1259947
and references cited therein. See for a recent review article. |
| 18. |
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315. doi:10.1021/acs.jmedchem.5b00258
and references cited therein. See for a recent review article. |
| 19. |
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422. doi:10.1021/acs.chemrev.5b00392
and references cited therein. See for a recent review article. |
| 20. |
Yerien, D. E.; Bonesi, S.; Postigo, A. Org. Biomol. Chem. 2016, 14, 8398. doi:10.1039/c6ob00764c
and references cited therein. See for a recent review article. |
| 22. | Liu, P.; Gao, Y.; Gu, W.; Shen, Z.; Sun, P. J. Org. Chem. 2015, 80, 11559. doi:10.1021/acs.joc.5b01961 |
| 28. | Pujari, S. A.; Kaliappan, K. P.; Valleix, A.; Grée, D.; Grée, R. Synlett 2008, 2503. doi:10.1055/s-2008-1078179 |
| 15. | Hudlicky, M., Ed. Chemistry of Organic Fluorine Compounds II: A Critical Review; ACS Monograph, Vol. 187; American Chemical Society: Washington, DC, 1995. |
| 16. | Kitazume, T.; Yamazaki, T. Experimental Methods in Organic Fluorine Chemistry; Gordon and Breach Science Publishers: Tokyo, 1998. |
| 23. | Monir, K.; Bagdi, A. K.; Ghosh, M.; Hajra, A. J. Org. Chem. 2015, 80, 1332. doi:10.1021/jo502928e |
| 14. | Casaubon, R. L.; Narayan, R.; Oalmann, C.; Vu, C. B. Substituted bicyclic azaheterocycles and analogues as sirtuin modulators. WO Patent WO2,013,059,587, Oct 19, 2012. |
| 30. | CCDC 1549632 (for compound 7a) and CCDC 1549633 (for compound 7e) contain the supplementary crystallographical data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre. |
| 13. | Moraski, G. C.; Markley, L. D.; Hipskind, P. A.; Boshoff, H.; Cho, S.; Franzblau, S. G.; Miller, M. J. ACS Med. Chem. Lett. 2011, 2, 466. doi:10.1021/ml200036r |
| 21. |
Hachem, A.; Grée, D.; Chandrasekhar, S.; Grée, R. Synthesis 2017, 49, 2101. doi:10.1055/s-0036-1589484
and references cited therein. |
| 26. | Nasr El Dine, A.; Khalaf, A.; Grée, D.; Tasseau, O.; Fares, F.; Jaber, N.; Lesot, P.; Hachem, A.; Grée, R. Beilstein J. Org. Chem. 2013, 9, 1943. doi:10.3762/bjoc.9.230 |
© 2017 Hariss et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)