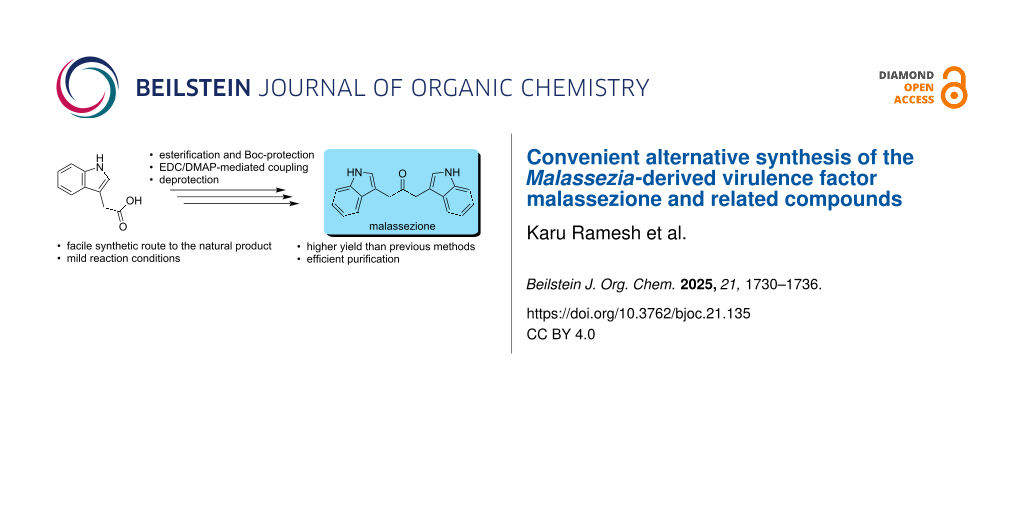
Abstract
Lipophilic yeasts of the genus Malassezia are commensal fungi that constitute the normal skin microbiota but may become pathogenic. These fungi, especially M. furfur, convert tryptophan into various alkaloid indoles such as malassezione, which may serve as virulence factors. To facilitate testing of malassezione as an aryl hydrocarbon receptor agonist and potential glucokinase activator, we developed a convenient synthetic route from commercially available indole-3-acetic acid. Treatment of the N-Boc-protected indole-3-acetic acid with N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDC) in the presence of DMAP generates the N,N′-Boc-protected malassezione, which upon deprotection yields malassezione in an overall yield of ca. 20%. This is an improvement over the preparation of the isonitrile followed by an Fe hydride initiated isonitrile–olefin intramolecular coupling reaction, which generated malassezione with an overall yield of ca. 5%. Furthermore, the present method may also be used to prepare related compounds.

Graphical Abstract
Introduction
Lipophilic yeasts of the genus Malassezia are commensal fungi that constitute the normal skin microbiota; however, when they become pathogenic, they are typically associated with various skin diseases, including pityriasis versicolor, malassezia folliculitis, atopic dermatitis, and seborrhoeic dermatitis in humans [1-7]. Additionally, Malassezia DNA has been suggested to play a role in the etiology in neurological disorders [7] and possibly other non-skin diseases [5]. These fungi, especially M. furfur, convert tryptophan into various alkaloid indoles such as malassezione (1), malassezin (2), which cyclizes to indolo[3,2-b]carbazole (3), other related indolo[3,2-b]carbazoles (4–7), pityriarubins (8–10), and others (11–19) (see Figure 1), which may serve as virulence factors [8-13]. Most significantly, some of these indoles have been shown to be potent aryl hydrocarbon receptor (AHR) ligands [8,9,14,15], which can lead to induction of melanocyte apoptosis and inhibition of neutrophil function [16,17].
![[1860-5397-21-135-1]](/bjoc/content/figures/1860-5397-21-135-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Various natural indole-containing compounds isolated from Malassezia furfur.
Figure 1: Various natural indole-containing compounds isolated from Malassezia furfur.
Malassezione (1, also referred to as malathidone [18]) is an AHR agonist [19,20]. Recently, in an effort to identify compounds as glucokinase activators to treat type 2 diabetes, structure-based virtual screening identified malassezione as a potential glucokinase activator [21]. Unfortunately, testing of malassezione is limited by the lack of a convenient synthesis. Indeed, beyond isolating the compound from cultured M. furfur [11,20], the only reported synthetic route to malassezione is via an iron-catalyzed Fukuyama-type indole synthesis, which afforded the compound in low yield [18]. Herein, we report an alternative synthesis from commercially available indole acetic acid, and demonstrate that the approach can be extended to related compounds.
Results and Discussion
Previously, malassezione had either been (i) isolated from cultures of M. furfur grown on ʟ-tryptophan as the sole nitrogen source followed by thin-layer chromatography and reversed-phase (RP)-HPLC to isolate and purify the compound from a complex mixture of indole alkaloids [11], (ii) prepared by aerobic incubation of indole-3-pyruvic acid at pH 7.4, 37 °C for 24 h followed by isolation and purification using RP-HPLC [20], or, more recently, (iii) prepared via an Fe hydride initiated isonitrile–olefin intramolecular coupling reaction (Scheme 1A) [18]. The reported synthetic route relies on an initial 4-step preparation of the isonitrile precursor, which was accomplished in an overall yield of ca. 14%. The subsequent iron-catalyzed reaction afforded 1 in only 35% yield. To avoid the low-yielding preparation of the isonitrile, we elected to use commercially available indole-3-acetic acid as the starting material, and to utilize an approach similar to that described for the synthesis of dibenzylketones from the DCC-induced condensation of phenylacetic acid reported by Bhandari and Ray [22] and others [23-27]. Initially, we converted indole-3-acetic acid to its methyl ester, followed by protection of the indole nitrogen with a Boc group and subsequent hydrolysis of the methyl ester to regenerate the carboxylic acid (Scheme 1B) in a manner similar to that described by Knör et al. [28]. Protection of the carboxylic acid group prior to conducting the N-Boc protection with di-tert-butyl dicarbonate ((Boc)2O) and DMAP was required to avoid competitive reaction of the carboxylic acid with (Boc)2O to form a tert-butyl ester, leading to unwanted side products and reduced selectivity. Once the acid was protected, the Boc group could be selectively introduced onto the indole nitrogen without side reactions. Overall, this sequence ensured clean N-Boc protection with minimal side reactions and easier purification. The N-Boc-protected indole-3-acetic acid was subsequently treated with N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDC) [29] in the presence of DMAP to generate the N,N′-Boc-protected malassezione (23). Use of EDC greatly facilitated the purification since the resulting urea derivative was easily removed with aqueous washes, thereby obviating the need for column purification to remove the more hydrophobic dicyclohexylurea obtained when DCC was employed to facilitate the reaction. Subsequent removal of the Boc groups with TFA furnished malassezione (1) in an overall yield of ca. 20% (4 steps) relative to an overall yield of ca. 5% via the preparation of the isonitrile followed by the Fe hydride initiated isonitrile–olefin intramolecular coupling reaction [18]. Protection of the indole nitrogens with the Boc group was preferable to the use of benzyl protecting groups since removal of the benzyl groups by hydrogenation was problematic (vide infra). The NMR spectral characteristics of the resulting material were identical to those reported for malassezione either isolated from cultures [11,20] or prepared from the isonitrile [18].
![[1860-5397-21-135-i1]](/bjoc/content/inline/1860-5397-21-135-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic routes to malassezione (1) from either A) an isonitrile precursor [18] or B) indole-3-acetic acid.
Scheme 1: Synthetic routes to malassezione (1) from either A) an isonitrile precursor [18] or B) indole-3-acetic ...
To further demonstrate the utility of the approach, we also prepared the dibenzylketone (25a) from phenylacetic acid in 80% yield (cf. 64% yield reported by Bhandari and Ray [22]) (Scheme 2). Previously, Bhandari and Ray demonstrated that the DCC-mediated reaction could be used with 4-methoxyphenylacetic acid to generate bis(4-methoxybenzyl) ketone, which could be converted to the corresponding bis(4-hydroxybenzyl) ketone (25c) by heating at 200 °C for 15 min to 3 h in the presence of pyridinium chloride [22,24]. Herein, we also show that the EDC-mediated coupling of 4-(benzyloxy)phenylacetic acid readily yields 25b, which may be debenzylated by Pd-catalyzed hydogenation to cleanly yield 25c. This affords a route to such compounds bearing hydroxylated aromatic groups that employs milder conditions than previously reported [22,24]. Finally, we prepared N,N′-dibenzylmalassezione (25d). Initially, this compound was prepared with the intent of using the benzyl group to protect the indole nitrogen rather than the Boc group. However, Pd-catalyzed hydrogenation of 25d led to a mixture of products, of which some were consistent with reduction of the indole ring.
With respect to the scalability of the synthesis, the reactions can be conducted on the gram-scale starting from indole-3-acetic acid with consistently good yields up to and including the EDC-mediated coupling step. However, the subsequent removal of the Boc protecting group proved more challenging. We observed lower yields during this step, despite screening a range of acids and conditions, including variations of the TFA treatment. Among the conditions tested, TFA gave the best results, though an optimal outcome was achieved only on the smaller, 100 mg scale. Scaling up the reaction at this stage increased the formation of side products. Consequently, it might be advisable to stockpile the penultimate synthetic intermediate 23 and subsequently conduct the deprotection on the smaller scale.
![[1860-5397-21-135-i2]](/bjoc/content/inline/1860-5397-21-135-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Various bis-substituted ketones prepared. a25c prepared from 25b.
Scheme 2: Various bis-substituted ketones prepared. a25c prepared from 25b.
Conclusion
Overall, we have developed a facile synthesis of the natural product malassezione, utilizing an EDC-mediated coupling of N-Boc-protected indole-3-acetic acid. The use of EDC permits easy removal of the water-soluble urea-based byproduct with an aqueous wash as opposed to the hydrophobic dicyclohexylurea arising when DCC is utilized, which is more difficult to separate from the hydrophobic malassezione. This route should afford ready access to malassezione for assessing its role as an AHR agonist [19,20] and potential glucokinase activator [21].
Experimental
General. Unless otherwise noted, all reagents were used as received from commercial sources: Sigma-Aldrich Canada Ltd. (Oakville, ON, Canada), TCI America (Portland, OR), or Fisher Scientific Canada (Ottawa, ON, Canada). Reactions were monitored using thin-layer chromatography on aluminium-backed silica plates (Sigma-Aldrich) using either UV-light (254 nm), iodine, KMnO4, phosphomolybdic acid, or p-anisaldehyde for visualization. Tetrahydrofuran (THF) was dried and distilled over sodium/benzophenone. Concentration under reduced pressure refers to the removal of solvent using a rotary evaporator, unless stated otherwise. All NMR spectra were obtained using a Bruker ASCEND 400 MHz spectrometer at the Nuclear Magnetic Resonance Research Resource (NMR-3, Dalhousie University). Chemical shifts (δ in ppm) for proton (1H) spectra are reported relative to the residual solvent signal for CDCl3 (δ 7.26) or methanol-d4 (δ 3.31) [30]. Chemical shifts (δ in ppm) for carbon (13C) spectra are reported relative to the residual solvent signal for CDCl3 (δ 77.16) or methanol-d4 (δ 49.00) [30]. Abbreviations used for reporting NMR spectral data are: bm, broad multiplet; bs, broad singlet; bt, broad triplet; d, doublet; dd, doublet of doublets; m, multiplet; q, quartet; s, singlet; and t, triplet. High-resolution (HR) electrospray ionization (ESI) mass spectra (MS) were collected using a Bruker microTOF Focus orthogonal ESI-TOF mass spectrometer instrument operating in either negative or positive ion mode as indicated in the experimental procedures (Chemistry Mass Spectrometry Core Facility, Dalhousie University). Melting points were recorded on Barnstead International (Model No. 1201D) melting point apparatus and are uncorrected.
tert-Butyl 3-(2-methoxy-2-oxoethyl)-1H-indole-1-carboxylate (21). A solution of indol-3-yl acetic acid (20, 5.1 g, 29.0 mmol, 1.0 equiv) in dry MeOH (200 mL) was cooled to 0 °C, and SOCl2 (10.5 mL, 145 mmol, 5.0 equiv) was added slowly. After stirring for 12 h at room temperature, the solution was concentrated under reduced pressure. The residue was taken up in EtOAc (200 mL) and subsequently washed with saturated aqueous NH4Cl (150 mL), saturated aqueous NaHCO3 (150 mL), and brine (150 mL). After drying over anhydrous MgSO4, the mixture was concentrated to dryness to give the pure methyl ester as a brown oil (5.1 g, 94%); Rf = 0.3 (EtOAc/hexane 1:2). Spectroscopic data for the methyl ester agreed with those in the literature [31]. The methyl ester (5.0 g, 26.0 mmol, 1.0 equiv) was then dissolved in acetonitrile (25 mL) and di-tert-butyl dicarbonate (17.0 g, 78 mmol, 3.0 equiv) and DMAP (0.63 g, 5.2 mmol, 0.2 equiv) were added [32]. The mixture was stirred for 2 h at room temperature after which the solvent was removed under reduced pressure. The residue was dissolved in EtOAc (250 mL) and subsequently washed with saturated aqueous NH4Cl (150 mL), saturated aqueous NaHCO3 (150 mL), and brine (100 mL). After drying over anhydrous MgSO4, the solvent was removed in vacuo and the crude product was purified by flash chromatography on silica gel (EtOAc/hexane; gradient 1:10 → 1:2) to give 21 as a colorless solid (6.7 g, 80%, 2 steps); Rf = 0.5 (EtOAc/hexane 1:2); mp 50–52 °C (lit. mp 53 °C [28]); 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.1 Hz, 1H, ArH), 7.58 (s, 1H, ArH), 7.54 (dt, J = 7.7 and 1.1 Hz, 1H, ArH), 7.34 (ddd, J = 8.4, 7.2 and 1.1 Hz, 1H, ArH), 7.26 (ddd, J = 8.4, 7.2 and 1.1 Hz, 1H, ArH), 3.73 (d, J = 1.1 Hz, 2H), 3.71 (s, 3H, CH3), 1.67 (s, 9H, 3 × CH3) ppm; 13C NMR (100 MHz, CDCl3) δ 171.5, 149.6, 135.5, 130.1, 124.6, 124.5, 122.7, 119.0, 115.3, 113.1, 83.6, 52.1, 30.9, 28.2 ppm. These spectroscopic data agreed with those in the literature [28].
2-(1-(tert-Butoxycarbonyl)-1H-indol-3-yl)acetic acid (22). A solution of lithium hydroxide (LiOH·H2O, 2.1 g, 51.8 mmol, 3 equiv) in H2O (250 mL) was added to a solution of 21 (5.0 g, 17.2 mmol, 1.0 equiv) in THF (300 mL) and MeOH (150 mL). After stirring for 18 h at room temperature, the mixture was concentrated in vacuo and a 10% aqueous solution of citric acid (100 mL) was added. The aqueous layer was extracted with EtOAc (3 × 50 mL) and the combined organic layers were washed with water (100 mL) and brine (100 mL), and then dried over anhydrous MgSO4. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (EtOAc/hexane gradient 1:10 → 1:1, 1% AcOH), yielding 22 as a colorless solid (3.7 g, 80%); Rf = 0.6 (MeOH/CHCl3 1:9, 1% AcOH); mp 116–118 °C (lit. mp 117–120 °C [28]); 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 8.3 Hz, 1H, ArH), 7.58 (s, 1H, ArH), 7.53 (dt, J = 7.8 and 1.1 Hz, 1H, ArH), 7.53 (ddd, J = 8.4, 7.2 and 1.3 Hz, 1H, ArH), 7.33 (ddd, J = 8.4, 7.2 and 1.3 Hz, 1H, ArH), 3.76 (s, J = 1.0 Hz, 2H), 1.66 (s, 9H, 3 × CH3) ppm; 13C NMR (100 MHz, CDCl3) δ 176.8, 149.7, 135.5, 135.0, 124.84, 124.80, 122.8, 119.1, 115.5, 112.5, 83.9, 30.9, 28.3 ppm; HRESIMS (m/z): [M + Na]+ calcd for C15H17NNaO4, 298.1050; found, 298.1057. These spectroscopic data agreed with those in the literature [28].
Di-tert-butyl 3,3'-(2-oxopropane-1,3-diyl)bis(1H-indole-1-carboxylate) (23). An oven-dried 50 mL flask equipped with a magnetic stir bar was charged with the Boc-protected acid 22 (1 g, 3.6 mmol, 1 equiv), EDC·HCl (1.3 g, 7.2 mmol, 2 equiv), and DMAP (87 mg, 0.7mmol, 0.2 equiv) in THF (10 mL) under an argon atmosphere. The reaction mixture was stirred at room temperature for 12 h. After completion of the reaction, the solvent was removed under reduced pressure. The crude reaction mixture was then diluted with EtOAc (100 mL) and washed with water (2 × 40 mL), followed by brine (40 mL). The organic layer was dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of EtOAc/hexane (1:10 → 1:1), affording compound 23 as a colorless viscous oil (425 mg, 50%); 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.3 Hz, 1H, ArH), 7.49 (s, 1H, ArH), 7.36–7.29 (m, 2H, ArH), 7.20 (td, J = 7.5 and 1.0 Hz, 1H, ArH), 3.85 (d, J = 1.0 Hz, 2H, CH2), 1.66 (s, 9H, 3 × CH3) ppm; 13C NMR (100 MHz, CDCl3) δ 205.0, 149.7, 135.5, 130.2, 124.8, 124.7, 122.8, 119.0, 115.4, 113.2, 83.8, 38.7, 28.3 ppm; HRESIMS (m/z): [M + Na]+ calcd for C29H32N2NaO5, 511.2203; found, 511.2222.
1,3-Di(1H-indol-3-yl)propan-2-one (1): The ketone 23 (200 mg, 0.4 mmol, 1 equiv) was dissolved in DCM (6 mL) in a 25 mL round bottom flask equipped with a magnetic stirrer. Trifluoroacetic acid (3 mL) was then added dropwise at 0 °C. The reaction was then stirred for 2 h as the temperature rose from 0 °C to room temperature. The solution was then concentrated in vacuo to yield an oily residue that was co-evaporated with diethyl ether (3 × 10 mL) under vacuum to produce the desired compound 1 as a beige viscous oil (70 mg, 60%); 1H NMR (400 MHz, CDCl3) δ 8.07 (s, 1H, NH), 7.47 (dd, J = 8.0 and 1.1 Hz, 1H, ArH(4/4′)), 7.35 (dt, J = 8.2 and 0.9 Hz, 1H, ArH(7/7′)), 7.20 (ddd, J = 8.2, 7.0 and 1.2 Hz, 1H, ArH(6/6′)), 7.11 (ddd, J = 8.0, 7.0 and 1.0 Hz, 1H, ArH(5/5′)), 7.05 (d, J = 2.4 Hz, 1H, ArH(2/2′)), 3.89 (s, 2H, CH2(8/8′)) ppm; 13C NMR (100 MHz, CDCl3) δ 207.2 (C9/9′), 136.3 (C7a/7a′), 127.5 (C3a/3a′), 123.4 (C2/2′), 122.4 (C6/6′), 119.9 (C5/5′), 118.9 (C4/4′), 113.4 (C7/7′), 108.9 (C3/3′), 38.7 (C8/8′) ppm; HRESIMS (m/z): [M + Na]+ calcd for C19H16N2NaO, 311.1155; found, 311.1163. These data are fully consistent with those published previously [20].
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-135-i3.svg?max-width=637&scale=1.18182)
The observation of 19 carbon atoms in 1 (HRESIMS) and 10 carbon signals in the 13C NMR spectrum is in accord with the molecule being symmetric. The structure of di-indole 1 was further supported by 2D NMR. The 1H-1H COSY spectrum showed couplings between the H4-H5, H5-H6, H6-H7, and H1-H2 protons, as well as long-range coupling between the H2-H8 protons. Again, these data are fully consistent with those published previously [20]. Together, these results indicate the presence of two indoles, one carbonyl, one methylene, and one methine group where the connectivities of these groups were supported from the 1H-13C HMBC spectrum. Couplings between the H8 and C9 (or H8′ and C9) atoms are consistent with the C8 and C8′ atoms linked to the carbonyl carbon (C9). In addition, couplings between the H8 and the C2, C3, C3a, and C8 carbon atoms, as well as between the H2 and the C3, C7a, and C8 carbon atoms, were observed. These couplings are consistent with the C8 and C8′ atoms being attached to the C3 and C3′ carbon atoms of the two indole groups, respectively. Furthermore, the 1H-13C HMBC spectrum showed couplings in the aromatic region consistent with the presence of the indole groups.
General procedure for the synthesis of ketones 25a,b, and d from acids 24a,b, and d. Adapting DCC/DMAP protocols reported previously [22-27], an oven-dried 50 mL flask equipped with a magnetic stir bar was charged with the acid (1 equiv, typically 1.0 g), EDC·HCl (2 equiv), and DMAP (0.2 equiv) in THF (10 mL) under an argon atmosphere. The reaction mixture was stirred at room temperature for 12 h. After completion of the reaction, the solvent was removed under reduced pressure, and the crude reaction mixture was diluted with EtOAc (100 mL), then washed with water (2 × 40 mL) and brine (40 mL). The organic layer was dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The resulting residue was subsequently purified by flash chromatography on silica gel using a hexane/ethyl acetate mixture as the eluent.
1,3-Diphenylpropan-2-one (25a). The reaction was conducted according to the general procedure using commercially available phenylacetic acid (1.0 g, 7.3 mmol, 1 equiv). The crude product was purified by chromatography on silica gel (hexane/EtOAc; 90:10) to afford 25a as a white solid (1.2 g, 80%); mp 108–110 °C; 1H NMR (400 MHz, CDCl3) δ 7.32–7.22 (m, 6H, ArH), 7.14–7.12 (m, 4H, ArH), 3.70 (s, 4H, 2 × CH2) ppm; 13C NMR (100 MHz, CDCl3) δ 205.8, 134.1, 129.6, 128.8, 127.2, 49.2 ppm. These spectroscopic data agreed with those in the literature [22,33,34].
1,3-Bis(4-(benzyloxy)phenyl)propan-2-one (25b). The reaction was conducted according to the general procedure using the commercially available 4-(benzyloxy)phenylacetic acid (1.0 g, 4.7 mmol, 1 equiv). The crude product was purified by chromatography on silica gel (hexane/EtOAc; 90:10) to afford 25b as a white solid (1.2 g, 75%); mp 110–112 °C (lit. mp 119 °C [35]); 1H NMR (400 MHz, CDCl3) δ 7.46–7.32 (m, 10H, ArH), 7.10–7.06 (m, 4H, ArH), 6.96–6.93 (m, 4H, ArH), 5.07 (s, 4H, 2 × OCH2), 3.66 (s, 4H, 2 X × CH2) ppm; 13C NMR (100 MHz, CDCl3) δ 206.5, 158.0, 137.1, 130.6, 128.7, 128.1, 127.6, 126.5, 115.2, 70.1, 48.2 ppm; HRESIMS (m/z): [M + Na]+ calcd for C29H26NaO3, 445.1774; found, 445.1777. These spectral data agreed with those in the literature [35].
1,3-Bis(4-hydroxyphenyl)propan-2-one (25c). An oven-dried 25 mL flask equipped with a magnetic stir bar was charged with 1,3-bis(4-(benzyloxy)phenyl)propan-2-one (25b, 100 mg, 0.2 mmol, 1 equiv) and dissolved in ethanol (3 mL). A catalytic amount of Pd/C was then added and hydrogenolysis was conducted for 4 h with stirring under an H2-filled balloon. The reaction was monitored by TLC and, after completion of the reaction, the mixture was filtered through Celite, followed by washing of the remaining Pd/C with ethanol (3 × 5 mL). The solvent was removed by evaporation under reduced pressure and the resulting crude compound was purified by chromatography on silica gel (hexane/EtOAc; 90:10) to afford 25c as a white solid (41 mg, 73%); mp 154–156 °C (lit. mp 164 °C [22]; 150–152 °C [35]; 157–158 °C [24]); 1H NMR (400 MHz, methanol-d4) δ 6.96–6.91 (m, 4H, ArH), 6.73–6.69 (m, 4H, ArH), 3.63 (s, 4H, 2 × CH2) ppm; 13C NMR (100 MHz, methanol-d4) δ 209.2, 156.8, 131.0, 125.8, 115.7, 47.2 ppm; HRESIMS (m/z): [M + Na]+ calcd for C15H14NaO3, 265.0835; found, 265.0842. These spectral data agreed with those in published reports [22,24,35].
1,3-Bis(1-benzyl-1H-indol-3-yl)propan-2-one (25d). Indole-3-acetic acid (1.0 g, 5.7 mmol) in DMF (10 mL) was cooled to 0 °C, and sodium hydride (273 mg, 11.4 mmol, 2 equiv) was added slowly with stirring. Benzyl bromide (1.9 g, 11.4 mmol, 2 equiv) was then added dropwise, and the reaction mixture was stirred for 12 h, allowing the temperature to gradually rise to room temperature. Upon completion of the reaction, the mixture was washed with cold LiCl solution (10%) and extracted with ethyl acetate (3 × 40 mL). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford 2-(1-benzyl-1H-indol-3-yl)acetic acid [36]. The resulting product was then used to synthesize ketone 25d following the general procedure. The crude product was purified by chromatography on silica gel (hexane/EtOAc; 90:10) to afford 25d as a yellow viscous oil (52 mg, 60%); 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 7.9 Hz, 1H, ArH), 7.30–7.22 (m, 4H, ArH), 7.17 (t, J = 7.6 Hz, 1H, ArH), 7.09 (dd, J = 7.7 and 2.4 Hz, 3H, ArH), 6.99 (s, 1H), 5.24 (s, 4H, 2 × CH2), 6.99 (s, 4H, 2 × CH2) ppm; 13C NMR (100 MHz, CDCl3) δ 207.2, 137.6, 136.7, 128.9, 127.8, 127.5, 127.0, 122.1, 119.6, 119.1, 109.9, 108.1, 50.1, 38.6 ppm; HRESIMS (m/z): [M + Na]+ calcd for C33H28N2NaO3, 491.2094; found, 491.2090.
Supporting Information
| Supporting Information File 1: Copies of NMR and MS spectra of synthesized compounds. | ||
| Format: PDF | Size: 2.3 MB | Download |
Data Availability Statement
All data that supports the findings of this study are available in the published article and/or the supporting information of this article.
References
-
Borelli, D.; Jacobs, P. H.; Nall, L. J. Am. Acad. Dermatol. 1991, 25, 300–305. doi:10.1016/0190-9622(91)70198-b
Return to citation in text: [1] -
Midgley, G. Med. Mycol. 2000, 38 (Suppl. 1), 9–16. doi:10.1080/mmy.38.s1.9.16
Return to citation in text: [1] -
Harada, K.; Saito, M.; Sugita, T.; Tsuboi, R. J. Dermatol. 2015, 42, 250–257. doi:10.1111/1346-8138.12700
Return to citation in text: [1] -
Prohic, A.; Jovovic Sadikovic, T.; Krupalija‐Fazlic, M.; Kuskunovic‐Vlahovljak, S. Int. J. Dermatol. 2016, 55, 494–504. doi:10.1111/ijd.13116
Return to citation in text: [1] -
Ianiri, G.; LeibundGut-Landmann, S.; Dawson, T. L. Annu. Rev. Microbiol. 2022, 76, 757–782. doi:10.1146/annurev-micro-040820-010114
Return to citation in text: [1] [2] -
Chang, C. H.; Stein, S. L. Pediatr. Derm. 2024, 41, 769–779. doi:10.1111/pde.15603
Return to citation in text: [1] -
Naik, B.; Sasikumar, J.; Das, S. P. Mol. Neurobiol. 2025, 62, 533–556. doi:10.1007/s12035-024-04270-w
Return to citation in text: [1] [2] -
Pindur, U.; Müller, J. Arch. Pharm. (Weinheim, Ger.) 1987, 320, 280–282. doi:10.1002/ardp.19873200317
Return to citation in text: [1] [2] -
Wille, G.; Mayser, P.; Thoma, W.; Monsees, T.; Baumgart, A.; Schmitz, H.-J.; Schrenk, D.; Polborn, K.; Steglich, W. Bioorg. Med. Chem. 2001, 9, 955–960. doi:10.1016/s0968-0896(00)00319-9
Return to citation in text: [1] [2] -
Krämer, H.-J.; Kessler, D.; Hipler, U.-C.; Irlinger, B.; Hort, W.; Bödeker, R.-H.; Steglich, W.; Mayser, P. ChemBioChem 2005, 6, 2290–2297. doi:10.1002/cbic.200500163
Return to citation in text: [1] -
Irlinger, B.; Bartsch, A.; Krämer, H.-J.; Mayser, P.; Steglich, W. Helv. Chim. Acta 2005, 88, 1472–1485. doi:10.1002/hlca.200590118
Return to citation in text: [1] [2] [3] [4] -
Hort, W.; Mayser, P. Curr. Opin. Infect. Dis. 2011, 24, 100–105. doi:10.1097/qco.0b013e328342f787
Return to citation in text: [1] -
Kurniadi, I.; Hendra Wijaya, W.; Timotius, K. H. Acta Dermatovenerol. Alp. Pannonica Adriat. 2022, 31, 65–70. doi:10.15570/actaapa.2022.8
Return to citation in text: [1] -
Krämer, H.-J.; Podobinska, M.; Bartsch, A.; Battmann, A.; Thoma, W.; Bernd, A.; Kummer, W.; Irlinger, B.; Steglich, W.; Mayser, P. ChemBioChem 2005, 6, 860–865. doi:10.1002/cbic.200400247
Return to citation in text: [1] -
Nadal Rodríguez, P.; Hartung, F.; Pedrola, M.; Coomar, S.; Diaz-Moreno, A.; Hätälä, A. M.; Rolfes, K. M.; Sánchez-Vera, I.; Gil, J.; Molins, E.; Viayna, A.; Hanzl, A.; Thomä, N. H.; Haarmann-Stemmann, T.; Luque, F. J.; Lavilla, R.; Ghashghaei, O. ACS Cent. Sci. 2025, 11, 629–641. doi:10.1021/acscentsci.5c00194
Return to citation in text: [1] -
Vlachos, C.; Schulte, B. M.; Magiatis, P.; Adema, G. J.; Gaitanis, G. Br. J. Dermatol. 2012, 167, 496–505. doi:10.1111/j.1365-2133.2012.11014.x
Return to citation in text: [1] -
Sparber, F.; LeibundGut-Landmann, S. Front. Immunol. 2017, 8, 1614. doi:10.3389/fimmu.2017.01614
Return to citation in text: [1] -
Zhang, T.; Yu, M.; Huang, H. Chem. Sci. 2021, 12, 10501–10505. doi:10.1039/d1sc03058b
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Nguyen, L. P.; Hsu, E. L.; Chowdhury, G.; Dostalek, M.; Guengerich, F. P.; Bradfield, C. A. Chem. Res. Toxicol. 2009, 22, 1897–1904. doi:10.1021/tx900043s
Return to citation in text: [1] [2] -
Chowdhury, G.; Dostalek, M.; Hsu, E. L.; Nguyen, L. P.; Stec, D. F.; Bradfield, C. A.; Guengerich, F. P. Chem. Res. Toxicol. 2009, 22, 1905–1912. doi:10.1021/tx9000418
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Malini, M.; Thilagavathi, R.; Kumar Singh, S.; Pravin, A.; Selvam, C. ChemistrySelect 2023, 8, e202204909. doi:10.1002/slct.202204909
Return to citation in text: [1] [2] -
Bhandari, S.; Ray, S. Synth. Commun. 1998, 28, 765–771. doi:10.1080/00032719808006472
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Resendiz, M. J. E.; Garcia-Garibay, M. A. Org. Lett. 2005, 7, 371–374. doi:10.1021/ol0480527
Return to citation in text: [1] [2] -
Toyoda, M.; Imai, Y.; Mori, T. J. Phys. Chem. Lett. 2017, 8, 42–48. doi:10.1021/acs.jpclett.6b02492
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Velasco-Rubio, Á.; Varela, J. A.; Saá, C. Org. Lett. 2020, 22, 3591–3595. doi:10.1021/acs.orglett.0c01053
Return to citation in text: [1] [2] -
Yao, L.; Hu, Q.; Bao, L.; Zhu, W.; Hu, Y. Org. Lett. 2021, 23, 4971–4975. doi:10.1021/acs.orglett.1c01304
Return to citation in text: [1] [2] -
González, J. M.; Vidal, X.; Ortuño, M. A.; Mascareñas, J. L.; Gulías, M. J. Am. Chem. Soc. 2022, 144, 21437–21442. doi:10.1021/jacs.2c09479
Return to citation in text: [1] [2] -
Knör, S.; Khrenov, A. V.; Laufer, B.; Saenko, E. L.; Hauser, C. A. E.; Kessler, H. J. Med. Chem. 2007, 50, 4329–4339. doi:10.1021/jm070304x
Return to citation in text: [1] [2] [3] [4] [5] -
Sheehan, J. C.; Cruickshank, P. A.; Boshart, G. L. J. Org. Chem. 1961, 26, 2525–2528. doi:10.1021/jo01351a600
Return to citation in text: [1] -
Gottlieb, H. E.; Kotlyar, V.; Nudelman, A. J. Org. Chem. 1997, 62, 7512–7515. doi:10.1021/jo971176v
Return to citation in text: [1] [2] -
Davies, H. M. L.; Townsend, R. J. J. Org. Chem. 2001, 66, 6595–6603. doi:10.1021/jo015617t
Return to citation in text: [1] -
Jasys, V. J.; Kelbaugh, P. R.; Nason, D. M.; Phillips, D.; Rosnack, K. J.; Saccomano, N. A.; Stroh, J. G.; Volkmann, R. A. J. Am. Chem. Soc. 1990, 112, 6696–6704. doi:10.1021/ja00174a037
Return to citation in text: [1] -
Tran, K.-V.; Bickar, D. J. Org. Chem. 2006, 71, 6640–6643. doi:10.1021/jo0607966
Return to citation in text: [1] -
Lee, B.; Chirik, P. J. J. Am. Chem. Soc. 2020, 142, 2429–2437. doi:10.1021/jacs.9b11944
Return to citation in text: [1] -
Diederich, F.; Carcanague, D. R. Helv. Chim. Acta 1994, 77, 800–818. doi:10.1002/hlca.19940770320
Return to citation in text: [1] [2] [3] [4] -
Li, F.; Chen, W.; Ai, Y.; Zhou, X.; Xiang, J.; Lu, H.; Dong, Y.; Yang, Q.; Zhang, J. J. Agric. Food Chem. 2024, 72, 21410–21418. doi:10.1021/acs.jafc.4c03536
Return to citation in text: [1]
| 30. | Gottlieb, H. E.; Kotlyar, V.; Nudelman, A. J. Org. Chem. 1997, 62, 7512–7515. doi:10.1021/jo971176v |
| 30. | Gottlieb, H. E.; Kotlyar, V.; Nudelman, A. J. Org. Chem. 1997, 62, 7512–7515. doi:10.1021/jo971176v |
| 31. | Davies, H. M. L.; Townsend, R. J. J. Org. Chem. 2001, 66, 6595–6603. doi:10.1021/jo015617t |
| 1. | Borelli, D.; Jacobs, P. H.; Nall, L. J. Am. Acad. Dermatol. 1991, 25, 300–305. doi:10.1016/0190-9622(91)70198-b |
| 2. | Midgley, G. Med. Mycol. 2000, 38 (Suppl. 1), 9–16. doi:10.1080/mmy.38.s1.9.16 |
| 3. | Harada, K.; Saito, M.; Sugita, T.; Tsuboi, R. J. Dermatol. 2015, 42, 250–257. doi:10.1111/1346-8138.12700 |
| 4. | Prohic, A.; Jovovic Sadikovic, T.; Krupalija‐Fazlic, M.; Kuskunovic‐Vlahovljak, S. Int. J. Dermatol. 2016, 55, 494–504. doi:10.1111/ijd.13116 |
| 5. | Ianiri, G.; LeibundGut-Landmann, S.; Dawson, T. L. Annu. Rev. Microbiol. 2022, 76, 757–782. doi:10.1146/annurev-micro-040820-010114 |
| 6. | Chang, C. H.; Stein, S. L. Pediatr. Derm. 2024, 41, 769–779. doi:10.1111/pde.15603 |
| 7. | Naik, B.; Sasikumar, J.; Das, S. P. Mol. Neurobiol. 2025, 62, 533–556. doi:10.1007/s12035-024-04270-w |
| 8. | Pindur, U.; Müller, J. Arch. Pharm. (Weinheim, Ger.) 1987, 320, 280–282. doi:10.1002/ardp.19873200317 |
| 9. | Wille, G.; Mayser, P.; Thoma, W.; Monsees, T.; Baumgart, A.; Schmitz, H.-J.; Schrenk, D.; Polborn, K.; Steglich, W. Bioorg. Med. Chem. 2001, 9, 955–960. doi:10.1016/s0968-0896(00)00319-9 |
| 14. | Krämer, H.-J.; Podobinska, M.; Bartsch, A.; Battmann, A.; Thoma, W.; Bernd, A.; Kummer, W.; Irlinger, B.; Steglich, W.; Mayser, P. ChemBioChem 2005, 6, 860–865. doi:10.1002/cbic.200400247 |
| 15. | Nadal Rodríguez, P.; Hartung, F.; Pedrola, M.; Coomar, S.; Diaz-Moreno, A.; Hätälä, A. M.; Rolfes, K. M.; Sánchez-Vera, I.; Gil, J.; Molins, E.; Viayna, A.; Hanzl, A.; Thomä, N. H.; Haarmann-Stemmann, T.; Luque, F. J.; Lavilla, R.; Ghashghaei, O. ACS Cent. Sci. 2025, 11, 629–641. doi:10.1021/acscentsci.5c00194 |
| 22. | Bhandari, S.; Ray, S. Synth. Commun. 1998, 28, 765–771. doi:10.1080/00032719808006472 |
| 20. | Chowdhury, G.; Dostalek, M.; Hsu, E. L.; Nguyen, L. P.; Stec, D. F.; Bradfield, C. A.; Guengerich, F. P. Chem. Res. Toxicol. 2009, 22, 1905–1912. doi:10.1021/tx9000418 |
| 8. | Pindur, U.; Müller, J. Arch. Pharm. (Weinheim, Ger.) 1987, 320, 280–282. doi:10.1002/ardp.19873200317 |
| 9. | Wille, G.; Mayser, P.; Thoma, W.; Monsees, T.; Baumgart, A.; Schmitz, H.-J.; Schrenk, D.; Polborn, K.; Steglich, W. Bioorg. Med. Chem. 2001, 9, 955–960. doi:10.1016/s0968-0896(00)00319-9 |
| 10. | Krämer, H.-J.; Kessler, D.; Hipler, U.-C.; Irlinger, B.; Hort, W.; Bödeker, R.-H.; Steglich, W.; Mayser, P. ChemBioChem 2005, 6, 2290–2297. doi:10.1002/cbic.200500163 |
| 11. | Irlinger, B.; Bartsch, A.; Krämer, H.-J.; Mayser, P.; Steglich, W. Helv. Chim. Acta 2005, 88, 1472–1485. doi:10.1002/hlca.200590118 |
| 12. | Hort, W.; Mayser, P. Curr. Opin. Infect. Dis. 2011, 24, 100–105. doi:10.1097/qco.0b013e328342f787 |
| 13. | Kurniadi, I.; Hendra Wijaya, W.; Timotius, K. H. Acta Dermatovenerol. Alp. Pannonica Adriat. 2022, 31, 65–70. doi:10.15570/actaapa.2022.8 |
| 23. | Resendiz, M. J. E.; Garcia-Garibay, M. A. Org. Lett. 2005, 7, 371–374. doi:10.1021/ol0480527 |
| 24. | Toyoda, M.; Imai, Y.; Mori, T. J. Phys. Chem. Lett. 2017, 8, 42–48. doi:10.1021/acs.jpclett.6b02492 |
| 25. | Velasco-Rubio, Á.; Varela, J. A.; Saá, C. Org. Lett. 2020, 22, 3591–3595. doi:10.1021/acs.orglett.0c01053 |
| 26. | Yao, L.; Hu, Q.; Bao, L.; Zhu, W.; Hu, Y. Org. Lett. 2021, 23, 4971–4975. doi:10.1021/acs.orglett.1c01304 |
| 27. | González, J. M.; Vidal, X.; Ortuño, M. A.; Mascareñas, J. L.; Gulías, M. J. Am. Chem. Soc. 2022, 144, 21437–21442. doi:10.1021/jacs.2c09479 |
| 22. | Bhandari, S.; Ray, S. Synth. Commun. 1998, 28, 765–771. doi:10.1080/00032719808006472 |
| 23. | Resendiz, M. J. E.; Garcia-Garibay, M. A. Org. Lett. 2005, 7, 371–374. doi:10.1021/ol0480527 |
| 24. | Toyoda, M.; Imai, Y.; Mori, T. J. Phys. Chem. Lett. 2017, 8, 42–48. doi:10.1021/acs.jpclett.6b02492 |
| 25. | Velasco-Rubio, Á.; Varela, J. A.; Saá, C. Org. Lett. 2020, 22, 3591–3595. doi:10.1021/acs.orglett.0c01053 |
| 26. | Yao, L.; Hu, Q.; Bao, L.; Zhu, W.; Hu, Y. Org. Lett. 2021, 23, 4971–4975. doi:10.1021/acs.orglett.1c01304 |
| 27. | González, J. M.; Vidal, X.; Ortuño, M. A.; Mascareñas, J. L.; Gulías, M. J. Am. Chem. Soc. 2022, 144, 21437–21442. doi:10.1021/jacs.2c09479 |
| 5. | Ianiri, G.; LeibundGut-Landmann, S.; Dawson, T. L. Annu. Rev. Microbiol. 2022, 76, 757–782. doi:10.1146/annurev-micro-040820-010114 |
| 20. | Chowdhury, G.; Dostalek, M.; Hsu, E. L.; Nguyen, L. P.; Stec, D. F.; Bradfield, C. A.; Guengerich, F. P. Chem. Res. Toxicol. 2009, 22, 1905–1912. doi:10.1021/tx9000418 |
| 28. | Knör, S.; Khrenov, A. V.; Laufer, B.; Saenko, E. L.; Hauser, C. A. E.; Kessler, H. J. Med. Chem. 2007, 50, 4329–4339. doi:10.1021/jm070304x |
| 7. | Naik, B.; Sasikumar, J.; Das, S. P. Mol. Neurobiol. 2025, 62, 533–556. doi:10.1007/s12035-024-04270-w |
| 18. | Zhang, T.; Yu, M.; Huang, H. Chem. Sci. 2021, 12, 10501–10505. doi:10.1039/d1sc03058b |
| 20. | Chowdhury, G.; Dostalek, M.; Hsu, E. L.; Nguyen, L. P.; Stec, D. F.; Bradfield, C. A.; Guengerich, F. P. Chem. Res. Toxicol. 2009, 22, 1905–1912. doi:10.1021/tx9000418 |
| 21. | Malini, M.; Thilagavathi, R.; Kumar Singh, S.; Pravin, A.; Selvam, C. ChemistrySelect 2023, 8, e202204909. doi:10.1002/slct.202204909 |
| 18. | Zhang, T.; Yu, M.; Huang, H. Chem. Sci. 2021, 12, 10501–10505. doi:10.1039/d1sc03058b |
| 28. | Knör, S.; Khrenov, A. V.; Laufer, B.; Saenko, E. L.; Hauser, C. A. E.; Kessler, H. J. Med. Chem. 2007, 50, 4329–4339. doi:10.1021/jm070304x |
| 19. | Nguyen, L. P.; Hsu, E. L.; Chowdhury, G.; Dostalek, M.; Guengerich, F. P.; Bradfield, C. A. Chem. Res. Toxicol. 2009, 22, 1897–1904. doi:10.1021/tx900043s |
| 20. | Chowdhury, G.; Dostalek, M.; Hsu, E. L.; Nguyen, L. P.; Stec, D. F.; Bradfield, C. A.; Guengerich, F. P. Chem. Res. Toxicol. 2009, 22, 1905–1912. doi:10.1021/tx9000418 |
| 11. | Irlinger, B.; Bartsch, A.; Krämer, H.-J.; Mayser, P.; Steglich, W. Helv. Chim. Acta 2005, 88, 1472–1485. doi:10.1002/hlca.200590118 |
| 28. | Knör, S.; Khrenov, A. V.; Laufer, B.; Saenko, E. L.; Hauser, C. A. E.; Kessler, H. J. Med. Chem. 2007, 50, 4329–4339. doi:10.1021/jm070304x |
| 18. | Zhang, T.; Yu, M.; Huang, H. Chem. Sci. 2021, 12, 10501–10505. doi:10.1039/d1sc03058b |
| 32. | Jasys, V. J.; Kelbaugh, P. R.; Nason, D. M.; Phillips, D.; Rosnack, K. J.; Saccomano, N. A.; Stroh, J. G.; Volkmann, R. A. J. Am. Chem. Soc. 1990, 112, 6696–6704. doi:10.1021/ja00174a037 |
| 16. | Vlachos, C.; Schulte, B. M.; Magiatis, P.; Adema, G. J.; Gaitanis, G. Br. J. Dermatol. 2012, 167, 496–505. doi:10.1111/j.1365-2133.2012.11014.x |
| 17. | Sparber, F.; LeibundGut-Landmann, S. Front. Immunol. 2017, 8, 1614. doi:10.3389/fimmu.2017.01614 |
| 11. | Irlinger, B.; Bartsch, A.; Krämer, H.-J.; Mayser, P.; Steglich, W. Helv. Chim. Acta 2005, 88, 1472–1485. doi:10.1002/hlca.200590118 |
| 20. | Chowdhury, G.; Dostalek, M.; Hsu, E. L.; Nguyen, L. P.; Stec, D. F.; Bradfield, C. A.; Guengerich, F. P. Chem. Res. Toxicol. 2009, 22, 1905–1912. doi:10.1021/tx9000418 |
| 28. | Knör, S.; Khrenov, A. V.; Laufer, B.; Saenko, E. L.; Hauser, C. A. E.; Kessler, H. J. Med. Chem. 2007, 50, 4329–4339. doi:10.1021/jm070304x |
| 18. | Zhang, T.; Yu, M.; Huang, H. Chem. Sci. 2021, 12, 10501–10505. doi:10.1039/d1sc03058b |
| 28. | Knör, S.; Khrenov, A. V.; Laufer, B.; Saenko, E. L.; Hauser, C. A. E.; Kessler, H. J. Med. Chem. 2007, 50, 4329–4339. doi:10.1021/jm070304x |
| 22. | Bhandari, S.; Ray, S. Synth. Commun. 1998, 28, 765–771. doi:10.1080/00032719808006472 |
| 33. | Tran, K.-V.; Bickar, D. J. Org. Chem. 2006, 71, 6640–6643. doi:10.1021/jo0607966 |
| 34. | Lee, B.; Chirik, P. J. J. Am. Chem. Soc. 2020, 142, 2429–2437. doi:10.1021/jacs.9b11944 |
| 29. | Sheehan, J. C.; Cruickshank, P. A.; Boshart, G. L. J. Org. Chem. 1961, 26, 2525–2528. doi:10.1021/jo01351a600 |
| 35. | Diederich, F.; Carcanague, D. R. Helv. Chim. Acta 1994, 77, 800–818. doi:10.1002/hlca.19940770320 |
| 35. | Diederich, F.; Carcanague, D. R. Helv. Chim. Acta 1994, 77, 800–818. doi:10.1002/hlca.19940770320 |
| 19. | Nguyen, L. P.; Hsu, E. L.; Chowdhury, G.; Dostalek, M.; Guengerich, F. P.; Bradfield, C. A. Chem. Res. Toxicol. 2009, 22, 1897–1904. doi:10.1021/tx900043s |
| 20. | Chowdhury, G.; Dostalek, M.; Hsu, E. L.; Nguyen, L. P.; Stec, D. F.; Bradfield, C. A.; Guengerich, F. P. Chem. Res. Toxicol. 2009, 22, 1905–1912. doi:10.1021/tx9000418 |
| 21. | Malini, M.; Thilagavathi, R.; Kumar Singh, S.; Pravin, A.; Selvam, C. ChemistrySelect 2023, 8, e202204909. doi:10.1002/slct.202204909 |
| 22. | Bhandari, S.; Ray, S. Synth. Commun. 1998, 28, 765–771. doi:10.1080/00032719808006472 |
| 24. | Toyoda, M.; Imai, Y.; Mori, T. J. Phys. Chem. Lett. 2017, 8, 42–48. doi:10.1021/acs.jpclett.6b02492 |
| 36. | Li, F.; Chen, W.; Ai, Y.; Zhou, X.; Xiang, J.; Lu, H.; Dong, Y.; Yang, Q.; Zhang, J. J. Agric. Food Chem. 2024, 72, 21410–21418. doi:10.1021/acs.jafc.4c03536 |
| 22. | Bhandari, S.; Ray, S. Synth. Commun. 1998, 28, 765–771. doi:10.1080/00032719808006472 |
| 24. | Toyoda, M.; Imai, Y.; Mori, T. J. Phys. Chem. Lett. 2017, 8, 42–48. doi:10.1021/acs.jpclett.6b02492 |
| 18. | Zhang, T.; Yu, M.; Huang, H. Chem. Sci. 2021, 12, 10501–10505. doi:10.1039/d1sc03058b |
| 24. | Toyoda, M.; Imai, Y.; Mori, T. J. Phys. Chem. Lett. 2017, 8, 42–48. doi:10.1021/acs.jpclett.6b02492 |
| 22. | Bhandari, S.; Ray, S. Synth. Commun. 1998, 28, 765–771. doi:10.1080/00032719808006472 |
| 22. | Bhandari, S.; Ray, S. Synth. Commun. 1998, 28, 765–771. doi:10.1080/00032719808006472 |
| 24. | Toyoda, M.; Imai, Y.; Mori, T. J. Phys. Chem. Lett. 2017, 8, 42–48. doi:10.1021/acs.jpclett.6b02492 |
| 35. | Diederich, F.; Carcanague, D. R. Helv. Chim. Acta 1994, 77, 800–818. doi:10.1002/hlca.19940770320 |
| 11. | Irlinger, B.; Bartsch, A.; Krämer, H.-J.; Mayser, P.; Steglich, W. Helv. Chim. Acta 2005, 88, 1472–1485. doi:10.1002/hlca.200590118 |
| 20. | Chowdhury, G.; Dostalek, M.; Hsu, E. L.; Nguyen, L. P.; Stec, D. F.; Bradfield, C. A.; Guengerich, F. P. Chem. Res. Toxicol. 2009, 22, 1905–1912. doi:10.1021/tx9000418 |
| 22. | Bhandari, S.; Ray, S. Synth. Commun. 1998, 28, 765–771. doi:10.1080/00032719808006472 |
| 18. | Zhang, T.; Yu, M.; Huang, H. Chem. Sci. 2021, 12, 10501–10505. doi:10.1039/d1sc03058b |
| 35. | Diederich, F.; Carcanague, D. R. Helv. Chim. Acta 1994, 77, 800–818. doi:10.1002/hlca.19940770320 |
© 2025 Ramesh and Bearne; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.