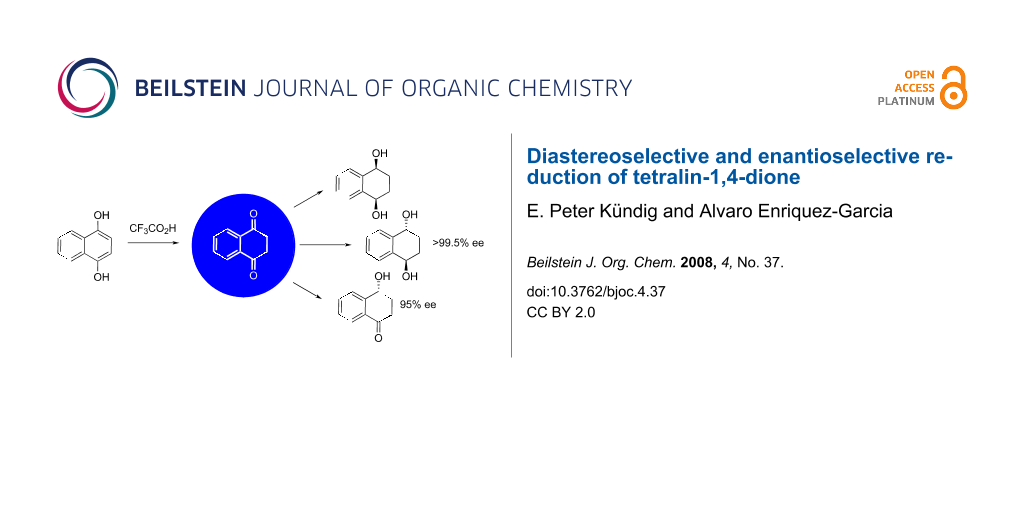
Abstract
Background
The chemistry of tetralin-1,4-dione, the stable tautomer of 1,4-dihydroxynaphthalene, has not been explored previously. It is readily accessible and offers interesting opportunities for synthesis.
Results
The title reactions were explored. L-Selectride reduced the diketone to give preferentially the cis-diol (d.r. 84 : 16). Red-Al gave preferentially the trans-diol (d.r. 13 : 87). NaBH4, LiAlH4, and BH3 gave lower diastereoselectivities (yields: 76–98%). Fractional crystallization allowed isolation of the cis-diol and the trans-diol (55% and 66% yield, respectively). Borane was used to cleanly give the mono-reduction product. Highly enantioselective CBS reductions afforded the trans-diol (72% yield, 99% ee) and the mono-reduction product (81%, 95% ee).
Conclusion
Diastereoselective and enantioselective reductions of the unexplored tetralin-1,4-dione provides a very convenient entry into a number of synthetically highly attractive 1,4-tetralindiols and 4-hydroxy-1-tetralone.

Graphical Abstract
Introduction
In this article, we briefly review synthetic approaches to 2,3-dihydro-1,4-naphthoquinone, more simply named tetralin-1,4-dione (2). This symmetric diketone is the stable tautomer of 1,4-dihydroxynaphthalene (1). Although known for many years, it has never been used in synthesis. The reactions of 2 that are reported in this article are those given in the title.
Tetralin-1,4-dione (2) is accessible by tautomerization, reduction, oxidation, and photolytic cycloreversion (Scheme 1 and Scheme 2). Tautomerization takes place upon melting 1 under an inert atmosphere or in a vacuum (>200 °C) [1-3]. The equilibrium mixture at this temperature consists of 1 and 2 in a ratio of 2 : 1 [3]. After cooling to ambient temperature, equilibration ceases and extracts with non-polar solvents are enriched with the more soluble 2. Tautomerization of 1 was also reported in trifluoroacetic acid, with 2 being the largely dominant species in solution [4].
![[1860-5397-4-37-i1]](/bjoc/content/inline/1860-5397-4-37-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Tautomerization of 1,4-dihydroxynaphthalene.
Scheme 1: Tautomerization of 1,4-dihydroxynaphthalene.
![[1860-5397-4-37-i2]](/bjoc/content/inline/1860-5397-4-37-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Alternative routes of access to tetralin-1,4-dione.
Scheme 2: Alternative routes of access to tetralin-1,4-dione.
Tetralin-1,4-dione (2) has also been obtained by catalytic hydrogenation of 1,4-naphthoquinone (3) using Wilkinson’s catalyst (70% yield) [5], by oxidation of 1-tetralone (4) with t-BuOOH and a dirhodium caprolactamate catalyst (27% yield at 29% conversion) [6], and by photolysis of the Dewar benzene 5 at low temperature in a solid matrix [7] (Scheme 2).
While dione 2 is readily synthesized, it remains a chemically unexplored curiosity. This simple molecule, and its π-metal complexes, drew our attention and interest for their potential in synthesis. Using the tautomerization of 1 in trifluoroacetic acid to generate 2 [4], we found that upon solvent evaporation the tautomer obtained was dihydroxynaphthalene 1, rather than diketone 2. During evaporation, the lower solubility of 1 led to its precipitation and this shifted the equilibrium back. This problem was solved by adding toluene to the mixture before evaporation under vacuum. This, and recrystallization (iPr2O) afforded 2 in 72% yield [8]. The straightforward route allowed the synthesis of gram quantities of 2 and the opportunity to study its uncharted chemistry.
This paper details the results of our studies of reductions of the carbonyl functions in diketone 2.
Results and Discussion
Diastereoselective bis-reduction of 2
Reduction of tetralin-1,4-dione (2) with a number of reducing agents afforded mixtures of diastereoisomeric cis-diol 6 and trans-diol 7 in the ratios shown in Table 1. It is important to mention here that these reactions do not occur when tautomer 1 is used.
Table 1: Diastereoselective reduction of tetralin-1,4-dione (2).
![[Graphic 1]](/bjoc/content/inline/1860-5397-4-37-i7.svg?max-width=637&scale=1.0)
|
|||
| Entry | Reducing agenta |
Ratiob
6 : 7 |
Yieldc |
|---|---|---|---|
| 1 | NaBH4 | 58 : 42 | 98% |
| 2 | LiAlH4 | 32 : 68 | 94% |
| 3 | Red-Al | 13 : 87 | 76% |
| 4 | BH3·THF | 61 : 39 | 93% |
| 5 | L-Selectride | 84 : 16 | 98% |
aSee Supporting Information File 1 for details. b 1H NMR ratios in DMSO-d6. cIsolated mixture of 6 and 7.
The reductions with NaBH4 (Table 1, entry 1) and BH3·THF (entry 4) gave the diols in high yields but with low diastereoselectivity, slightly favoring the cis-diastereoisomer 6. In contrast, reduction with LiAlH4 (entry 2) and, more pronounced, with [Al(H2)(OCH2CH2OMe)2][Na] (Red-Al) favored the trans-diastereoisomer 7. Fractional crystallization of the 13 : 87 mixture (entry 3) afforded pure 7 in 55% yield. The reason for the diastereoselectivity in this reaction may have its origin in the delivery of the second hydride from the same aluminium moiety (Scheme 3). Conversely, lithium tri-sec-butylborohydride (L-selectride) afforded a product enriched with cis-tetralin-1,4-diol (6) (entry 5). The high diastereoselectivity presumably is a consequence of the bulky reducing agent. Following the first reduction and formation of the 4-(boranyloxy)-1-tetralone, addition of a second equivalent of L-selectride would be expected to occur from the less hindered face. Hydrolysis then yields preferentially the cis-diastereoisomer 6. The ca. 5 : 1 mixture of 6 and 7 could not be efficiently separated by flash chromatography but recrystallization from iPr2O gave cis-1,4-dihydroxytetralin (6) in 66% yield.
![[1860-5397-4-37-i3]](/bjoc/content/inline/1860-5397-4-37-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed origin of diastereomeric preference in the reduction of 2.
Scheme 3: Proposed origin of diastereomeric preference in the reduction of 2.
Diols 6 and 7 have been reported previously. They were obtained by treatment of tetralin with NBS to give a 1 : 1 mixture of the corresponding cis and trans-dibromides, which were converted into diacetates with AgOAc (81% yield). Saponification and fractional recrystallization from MeOH / Et2O then afforded pure 6 and 7 though isolated yields were not reported [9]. The meso-diol 6 has been used as substrate in enantioselective oxidation [10] and in asymmetric acylation [9,11].
We conclude that while conditions for an efficient highly diastereoselective one-step reduction of both carbonyl functions in 2 have not been realized, enrichment of one or the other diastereoisomer by choice of reducing agent is feasible and acceptable yields of pure diastereoisomers can be obtained.
Enantioselective bis-reduction of 2
Asymmetric reduction of dione 2 was probed next. This was carried out successfully as shown in Scheme 4 and gave, after two recrystallizations from diisopropylether, (−)-(1R,4R)-tetralin-1,4-diol (R,R-7) in 72% yield and 99% ee [8]. Only small amounts (ca. 7%) of the cis stereoisomer 6 were detected by 1H NMR in the crude product. The synthesis of diol 7 in highly enantiomerically enriched form is thus easier than that of the racemate.
![[1860-5397-4-37-i4]](/bjoc/content/inline/1860-5397-4-37-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Enantioselective reduction of 2.
Scheme 4: Enantioselective reduction of 2.
The absolute configuration of (−)-(1R,4R)-7 agrees with the reliable stereochemical model for the CBS reduction. To our knowledge there is no viable published alternative synthetic access to this C2 symmetric chiral diol. Compound (−)-(1R,4R)-7 was previously obtained by HPLC separation of a 1 : 1 mixture of the cis- and trans-diols obtained in 55% yield from a four step sequence from (R)-1-tetralol [12].
Mono-reduction of 2
Mono-reduction was achieved with a reduced amount of borane compared to the reduction detailed above. For the bis reduction, a molar ratio of 2 / BH3 of 0.83 was used. Adjusting the ratio to 2.2 (see experimental part) afforded rac-9 in good yield (Scheme 5).
![[1860-5397-4-37-i5]](/bjoc/content/inline/1860-5397-4-37-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
The high yield in mono-reduction is in accord with the expected higher reactivity of the dione 2 compared to the mono-ketone 9.
Enantioselective mono-reduction of 2
With an efficient protocol for the synthesis of rac-9 and of R,R-7 in hand, research then focused on the more challenging task of enantioselective mono-reduction. First, CBS reduction was performed by slow (1 h) addition of dione 2 to a solution of BH3·THF (0.45 equiv) and catalyst 8. However, background reduction by BH3·THF was competitive under these conditions and while (−)-(4R)-4-hydroxy-1-tetralone (R-9) could be isolated in 93% yield, its enantiomeric excess was a modest 53% ee.
A way to achieve a high ee in mono-reduction was via 1-trimethylsiloxy-4-oxotetralin-1-carbonitrile (10) as protected equivalent of dione 2. Slow addition over 2 h of a THF solution of ketone 10 to a solution of BH3·THF (0.6 equiv) and catalyst 8 in THF at −30 °C gave, after MeOH quenching and TBAF deprotection, (−)-9 in 85% yield and 95% ee (Scheme 6).
![[1860-5397-4-37-i6]](/bjoc/content/inline/1860-5397-4-37-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Enantioselective mono-reduction of 2.
Scheme 6: Enantioselective mono-reduction of 2.
Cyanohydrin silylether 10 partially hydrolyzes on silica and, as it turned out, isolation of this intermediate is not required and this provided a reliable and efficient sequence to highly enantiomerically enriched 9 (Scheme 5). In the course of this optimization, we also isolated cyanohydrin 11.
We note literature precedent for procedures for the asymmetric synthesis of 9. The first involves as the key step kinetic resolution by enzymatic hydrolysis of the corresponding acetate with porcine pancreatic lipase giving (−)-9 in 47% yield and 95% ee [13]. A second approach uses a Pd-catalyzed asymmetric oxidation of meso-tetralin-1,4-diol (6) with (−)-sparteine (20 mol %) to give (+)-9 in 72% yield and 95% ee [10].
We note that chiral 1,4-disubstituted tetralins are of interest in medicinal chemistry. An example is the commercial antidepressant drug sertraline (Zoloft ®) [14-16]. A number of natural products such as preussomerin A [17], catalponol [18], junglanoside A [19], and isoshinanolone [20] contain the 4-hydroxy-1-tetralone unit. 4-Hydroxy-1-tetralone (9) itself is a naturally occurring compound isolated from Ampelocera edentula with activity against cutaneous leishmaniasis [21]. The straightforward access to highly enantiomerically enriched 9 reported here will be useful.
Conclusion
Diastereoselective and enantioselective reductions of the unexplored tetralin-1,4-dione provides a very convenient entry into a number of synthetically highly attractive 1,4-tetralindiols and 4-hydroxy-1-tetralone.
Supporting Information
| Supporting Information File 1: Experimental procedures, full spectroscopic and analytical data of compounds 2, 6, 7, 9–11. | ||
| Format: DOC | Size: 263.5 KB | Download |
References
-
Madinaveitia, A.; Olay, E. An. Soc. Esp. Fis. Quim. 1933, 31, 134–138.
Return to citation in text: [1] -
Thomson, R. H. J. Chem. Soc. 1950, 1737–1742. doi:10.1039/JR9500001737
Return to citation in text: [1] -
Pearson, M. S.; Jensky, B. J.; Greer, F. X.; Hagstrom, J. P.; Wells, N. M. J. Org. Chem. 1978, 43, 4617–4622. doi:10.1021/jo00418a017
Return to citation in text: [1] [2] -
Laatsch, H. Liebigs Ann. Chem. 1980, 140–157. doi:10.1002/jlac.198019800111
Return to citation in text: [1] [2] -
Birch, A. J.; Walker, K. A. M. Tetrahedron Lett. 1967, 3457–3458. doi:10.1016/S0040-4039(01)89820-8
Return to citation in text: [1] -
Catino, A. J.; Nichols, J. M.; Choi, H.; Gottipamula, S.; Doyle, M. P. Org. Lett. 2005, 7, 5167–5170. doi:10.1021/ol0520020
Return to citation in text: [1] -
Tsuji, T.; Okuyama, M.; Ohkita, M.; Kawai, H.; Suzuki, T. J. Am. Chem. Soc. 2003, 125, 951–961. doi:10.1021/ja021206y
Return to citation in text: [1] -
Kündig, E. P.; García, A. E.; Lomberget, T.; Bernardinelli, G. Angew. Chem., Int. Ed. 2006, 45, 98–101. doi:10.1002/anie.200502588
Return to citation in text: [1] [2] -
Yamada, S.; Katsumata, H. J. Org. Chem. 1999, 64, 9365–9373. doi:10.1021/jo990892p
Return to citation in text: [1] [2] -
Ferreira, E. M.; Stoltz, B. M. J. Am. Chem. Soc. 2001, 123, 7725–7726. doi:10.1021/ja015791z
Return to citation in text: [1] [2] -
Yamada, S.; Misono, T.; Iwai, Y.; Masumizu, A.; Akiyama, Y. J. Org. Chem. 2006, 71, 6872–6880. doi:10.1021/jo060989t
Return to citation in text: [1] -
Boyd, D. R.; Sharma, N. D.; Kerley, N. A.; McConville, G.; Allen, C. C. R.; Blacker, A. J. ARKIVOC 2003, No. vii, 32–48.
Return to citation in text: [1] -
Joly, S.; Nair, M. S. Tetrahedron: Asymmetry 2001, 12, 2283–2287. doi:10.1016/S0957-4166(01)00404-9
Return to citation in text: [1] -
Williams, M.; Quallich, G. J. Chem. Ind. 1990, 315.
Return to citation in text: [1] -
Quallich, G. J. Chirality 2005, 17, S120–S126. doi:10.1002/chir.20113
Return to citation in text: [1] -
Lautens, M.; Rovis, T. Tetrahedron 1999, 55, 8967–8976. doi:10.1016/S0040-4020(99)00456-1
Return to citation in text: [1] -
Quesada, E.; Stockley, M.; Ragot, J. P.; Prime, M. E.; Whitwood, A. C.; Taylor, R. J. K. Org. Biomol. Chem. 2004, 2, 2483–2495. doi:10.1039/b407895k
Return to citation in text: [1] -
McDaniel, C. A. J. Chem. Ecol. 1992, 18, 359–369. doi:10.1007/BF00994237
Return to citation in text: [1] -
Liu, L.; Li, W.; Koike, K.; Zhang, S.; Nikaido, T. Chem. Pharm. Bull. 2004, 52, 566–569. doi:10.1248/cpb.52.566
Return to citation in text: [1] -
Anh, N. H.; Ripperger, H.; Porzel, A.; Sung, T. V.; Adam, G. Phytochemistry 1997, 44, 549–551. doi:10.1016/S0031-9422(96)00510-9
Return to citation in text: [1] -
Fournet, A.; Angelo Barrios, A.; Muñoz, V.; Hocquemiller, R.; Roblot, F.; Cavé, A. Planta Med. 1994, 60, 8–12. doi:10.1055/s-2006-959397
Return to citation in text: [1]
| 19. | Liu, L.; Li, W.; Koike, K.; Zhang, S.; Nikaido, T. Chem. Pharm. Bull. 2004, 52, 566–569. doi:10.1248/cpb.52.566 |
| 17. | Quesada, E.; Stockley, M.; Ragot, J. P.; Prime, M. E.; Whitwood, A. C.; Taylor, R. J. K. Org. Biomol. Chem. 2004, 2, 2483–2495. doi:10.1039/b407895k |
| 1. | Madinaveitia, A.; Olay, E. An. Soc. Esp. Fis. Quim. 1933, 31, 134–138. |
| 2. | Thomson, R. H. J. Chem. Soc. 1950, 1737–1742. doi:10.1039/JR9500001737 |
| 3. | Pearson, M. S.; Jensky, B. J.; Greer, F. X.; Hagstrom, J. P.; Wells, N. M. J. Org. Chem. 1978, 43, 4617–4622. doi:10.1021/jo00418a017 |
| 6. | Catino, A. J.; Nichols, J. M.; Choi, H.; Gottipamula, S.; Doyle, M. P. Org. Lett. 2005, 7, 5167–5170. doi:10.1021/ol0520020 |
| 10. | Ferreira, E. M.; Stoltz, B. M. J. Am. Chem. Soc. 2001, 123, 7725–7726. doi:10.1021/ja015791z |
| 5. | Birch, A. J.; Walker, K. A. M. Tetrahedron Lett. 1967, 3457–3458. doi:10.1016/S0040-4039(01)89820-8 |
| 14. | Williams, M.; Quallich, G. J. Chem. Ind. 1990, 315. |
| 15. | Quallich, G. J. Chirality 2005, 17, S120–S126. doi:10.1002/chir.20113 |
| 16. | Lautens, M.; Rovis, T. Tetrahedron 1999, 55, 8967–8976. doi:10.1016/S0040-4020(99)00456-1 |
| 12. | Boyd, D. R.; Sharma, N. D.; Kerley, N. A.; McConville, G.; Allen, C. C. R.; Blacker, A. J. ARKIVOC 2003, No. vii, 32–48. |
| 3. | Pearson, M. S.; Jensky, B. J.; Greer, F. X.; Hagstrom, J. P.; Wells, N. M. J. Org. Chem. 1978, 43, 4617–4622. doi:10.1021/jo00418a017 |
| 13. | Joly, S.; Nair, M. S. Tetrahedron: Asymmetry 2001, 12, 2283–2287. doi:10.1016/S0957-4166(01)00404-9 |
| 9. | Yamada, S.; Katsumata, H. J. Org. Chem. 1999, 64, 9365–9373. doi:10.1021/jo990892p |
| 9. | Yamada, S.; Katsumata, H. J. Org. Chem. 1999, 64, 9365–9373. doi:10.1021/jo990892p |
| 11. | Yamada, S.; Misono, T.; Iwai, Y.; Masumizu, A.; Akiyama, Y. J. Org. Chem. 2006, 71, 6872–6880. doi:10.1021/jo060989t |
| 8. | Kündig, E. P.; García, A. E.; Lomberget, T.; Bernardinelli, G. Angew. Chem., Int. Ed. 2006, 45, 98–101. doi:10.1002/anie.200502588 |
| 8. | Kündig, E. P.; García, A. E.; Lomberget, T.; Bernardinelli, G. Angew. Chem., Int. Ed. 2006, 45, 98–101. doi:10.1002/anie.200502588 |
| 20. | Anh, N. H.; Ripperger, H.; Porzel, A.; Sung, T. V.; Adam, G. Phytochemistry 1997, 44, 549–551. doi:10.1016/S0031-9422(96)00510-9 |
| 7. | Tsuji, T.; Okuyama, M.; Ohkita, M.; Kawai, H.; Suzuki, T. J. Am. Chem. Soc. 2003, 125, 951–961. doi:10.1021/ja021206y |
| 10. | Ferreira, E. M.; Stoltz, B. M. J. Am. Chem. Soc. 2001, 123, 7725–7726. doi:10.1021/ja015791z |
| 21. | Fournet, A.; Angelo Barrios, A.; Muñoz, V.; Hocquemiller, R.; Roblot, F.; Cavé, A. Planta Med. 1994, 60, 8–12. doi:10.1055/s-2006-959397 |
© 2008 Kündig and Enriquez-Garcia; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)