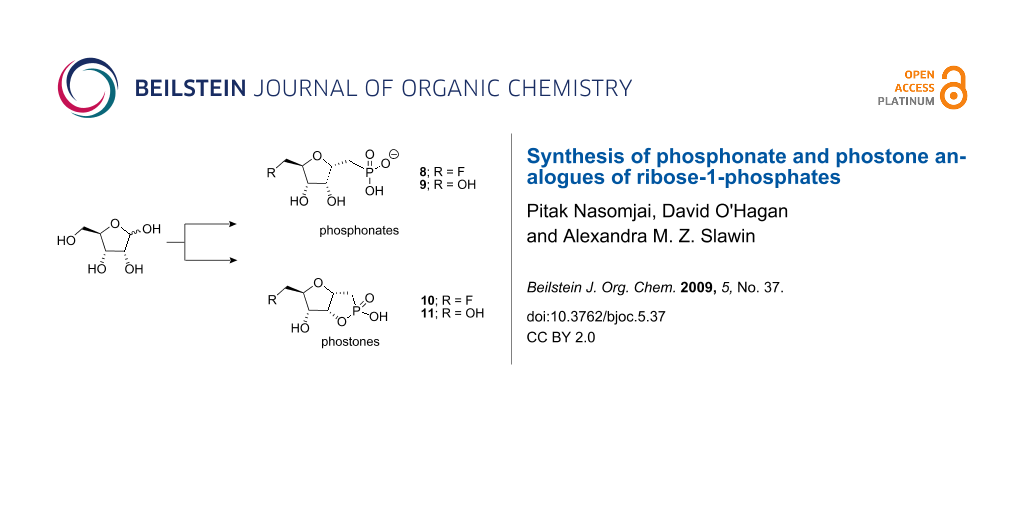
Abstract
The synthesis of phosphonate analogues of ribose-1-phosphate and 5-fluoro-5-deoxyribose-1-phosphate is described. Preparations of both the α- and β-phosphonate anomers are reported for the ribose and 5-fluoro-5-deoxyribose series and a synthesis of the corresponding cyclic phostones of each α-ribose is also reported. These compounds have been prepared as tools to probe the details of fluorometabolism in S. cattleya.

Graphical Abstract
Introduction
Fluoroacetate (1) and 4-fluorothreonine (2) are unusual secondary metabolites in that they contain a fluorine atom. They are elaborated by the bacterium Streptomyces cattleya as part of its defense strategy since 4-fluorothreonine (2) has antibiotic activity and fluoroacetate (1) is a toxin [1]. The biosynthetic pathway from fluoride ion to these fluorometabolites has largely been elucidated and is summarised in Scheme 1 [2].
![[1860-5397-5-37-i1]](/bjoc/content/inline/1860-5397-5-37-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Biosynthetic pathway from fluoride ion to fluoroacetate 1 and 4-fluorothreonine 2 in S. cattleya [2].
Scheme 1: Biosynthetic pathway from fluoride ion to fluoroacetate 1 and 4-fluorothreonine 2 in S. cattleya [2].
The first committed step in the biosynthesis involves the fluorinase, an enzyme that catalyses nucleophilic attack of fluoride ion on SAM (3) to generate 5′-FDA (4). The initial fluorinated product 5′-FDA (4) is then depurinated by a purine nucleotide phosphorylase (PNP) to give 5-fluoro-5-deoxyribose-1-phosphate (5). This phosphate intermediate becomes the substrate for a ring opening isomerisation reaction converting 5-fluoro-5-deoxyribose-1-phosphate (5) to 5-fluoro-5-deoxyribulose-1-phosphate (6). Further processing to fluoroacetaldehyde (7) and then conversion to fluoroacetate (1) and 4-fluorothreonine (2) complete the pathway. The focus of this research involved generating a potential inhibitor of either the PNP or the subsequent isomerase enzymes as a tool, to try to accumulate biosynthesis intermediates in cell free extract studies [3]. We have already demonstrated that the S. cattleya PNP can depurinate both 5′-FDA and adenosine, and thus the presence or absence of fluorine at the C-5′-position is not a requisite for catalysis. The next enzymatic step involves a ring opening isomerisation of the ribose-1-phosphate 5. In this paper we report the synthesis of phosphonate analogues 8 and 9 of ribose-1-phosphate 5 where the linking phosphate oxygen has been replaced by a methylene group and for 9 the fluorine has been replaced by OH (see Figure 1). These phosphonates were considered as candidate inhibitors for either the PNP or the isomerase enzyme, with potential utility as tools to interrogate the biosynthesis system, and as inert candidate substrate analogues for enzyme co-crystallisation studies. We also report the synthesis of the cyclic phostone analogues 10 and 11 (Figure 1), which were viewed also as potential substrate analogues for co-crystallisation studies, particularly with the isomerase enzyme which converts 5 to 6 during the biosynthesis.
![[1860-5397-5-37-1]](/bjoc/content/figures/1860-5397-5-37-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Synthesized phosphonates 8 and 9 and cyclic phostones 10 and 11.
Figure 1: Synthesized phosphonates 8 and 9 and cyclic phostones 10 and 11.
Results and Discussion
The target phosphonates 8 and 9 have been prepared by routes A and B as shown in Scheme 1 and Scheme 2 respectively. The routes start either from D-ribonic-γ-lactone (12) or D-ribose (13). For each route the key synthetic steps are the same involving installation of the dimethyl phosphonate moiety on a 2,3-O-isopropylidene ribofuranoside via a Wadsworth-Emmons reaction [4]. Route A (Scheme 2) started with acetonide protection of D-ribonic-γ-lactone (12) to generate 2,3-isopropylidene 14 [5]. Fluorination of the free 5-hydroxyl group with Deoxo-fluor™ [6] gave 5-deoxy-5-fluoro-2,3-O-isopropylidene-D-ribonic-γ-lactone (15) in good yield and then subsequent reduction of lactone 15 with NaBH4 resulted in the fully reduced diol 16. Diol 16 has been previously synthesised [7], however this route appears to offer a more efficient preparation. In our case, even with limiting NaBH4, mixtures of diol 16 and 5-deoxy-5-fluoro-2,3-ribofuranoside 17 were obtained, however treatment with DIBAL-H at 40 °C [8] gave a clean reaction and 5-fluororibofuranoside 17 was recovered in moderate yield. Finally a Wadsworth-Emmons reaction [4] of 17 using tetramethyl methylenediphosphonate in DCM/aq. NaOH gave a smooth conversion to 5-fluorophosphonate 18 which had an α:β ratio of 3.2:1. (The major α-anomer was assigned for 18 based on the subsequent NMR assignments of 23a and b, see below.) These diastereoisomers were inseparable by conventional chromatography.
![[1860-5397-5-37-i2]](/bjoc/content/inline/1860-5397-5-37-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic route A. Reagents and conditions: a) acetone, concd H2SO4 (cat.), 6 h, 86%; b) Deoxo-fluor™, DCM, 40 °C, 30 min, 84%; c) NaBH4, ethanol, 0 °C, 2 h, 65%; d) DIBAL-H, toluene, 40 °C, 64%; e) CH2[P(O)(OMe)2]2, DCM, 50% aq NaOH, 60%.
Scheme 2: Synthetic route A. Reagents and conditions: a) acetone, concd H2SO4 (cat.), 6 h, 86%; b) Deoxo-fluo...
Due to the inability to separate α−18 from β−18, route B (Scheme 3) was explored as an alternative. Following the protocol demonstrated by Meyer et al. [4], 5-O-trityl-phosphonates 21a and 21b were obtained as individual epimers after chromatographic separation of the initial product mixture (α:β of 4:1). Detritylation of 21a, b was accomplished with ZnCl2 [9] to generate the 5-hydroxyphosphonates 22a and 22b in a good yield. The free alcohol of both epimers was then fluorinated at the 5-position using tosylfluoride and TBAF in refluxing THF [10,11]. Sequential deprotection of 23a and 23b with TMSBr and then TFA yielded the free phosphonic acids which were neutralised with cyclohexylamine [12] to generate the non-hygroscopic salts 8a, b and 9a, b which could be recrystallised from MeOH/acetone. X-Ray structure analysis of a suitable crystal of the fluoromethyl phosphonate 8a was obtained (Figure 2) which confirmed its structure and stereochemical relationship to intermediate ribose-1-phosphate metabolite 5. Clearly, phosphonate 8a formed a 1:1 salt with the amine. Additionally, 23a and 22a were treated in separate reactions with acetic anhydride in dry pyridine [13] to generate the cyclic phostones 10 and 11 respectively. In each case this conversion could be conveniently followed by a change in the 31P-NMR resonance of 27 ppm (phosphonate) to 44 ppm, characteristic of a cyclic phostone. Purification by chromatography allowed the recovery of the phostones 10 and 11 in moderate yield.
![[1860-5397-5-37-i3]](/bjoc/content/inline/1860-5397-5-37-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthetic route B. Reagents and conditions: a) acetone, concd H2SO4 (cat.), 4 h, 90%; b) TrCl, pyridine, DMAP, rt, 16 h, 94%; c) CH2[P(O)(OMe)2]2, 50% aq NaOH; DCM, 64%; d) anhyd ZnCl2, DCM, rt, 2 h, 78–81%; e) TsF, TBAF, THF, reflux, 16 h, 83–92%; f) i) TMSBr, DCM, rt; ii) TFA:H2O (1:1), iii) cyclohexylamine, pH ~11, 84–87%; g) i) TMSBr, DCM, rt; ii) TFA:H2O (1:1), iii) Ac2O, pyridine, 16 h, 40–47%.
Scheme 3: Synthetic route B. Reagents and conditions: a) acetone, concd H2SO4 (cat.), 4 h, 90%; b) TrCl, pyri...
![[1860-5397-5-37-2]](/bjoc/content/figures/1860-5397-5-37-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-Ray crystal structure of cyclohexylammonium salt 8a.
Figure 2: X-Ray crystal structure of cyclohexylammonium salt 8a.
Incubation of the phosphonate salts 8a and 9a with the S. cattleya PNP did not result in an inhibitory effect, and clearly the phosphonates are not good substrate analogues of the phosphates with this enzyme. We are currently exploring phosphonates 8a and 9a as inhibitors of the isomerase, the next enzyme on the pathway, and in order to probe the molecular mechanism of this enzyme, these phosphonates and the phostones 10 and 11 are being used in co-crystallisation trials with each of the over-expressed enzymes (PNP and isomerase).
Experimental
Nuclear magnetic resonance (NMR) spectra were obtained using Bruker Advance 300 and Bruker Advance II 400. All chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) are given in Hertz (Hz). Melting points were determined in Pyrex capillaries using a Gallenkamp Griffin MPA350.BM2.5 melting point apparatus. Infra red (IR) spectra were recorded on Nicolet Avatar 360 FT-IR. High resolution mass spectrometry (HRMS) was carried out using a Micromass LCT (Manchester, UK) mass spectrometer with electrospray ionization (ESI) operating in positive and negative modes. Optical rotations were measured on Perkin-Elmer model 341 polarimeter.
5-Deoxy-5-fluoro-2,3-O-isopropylidene-D-ribono-1,4-lactone (15). Deoxo-Fluor™ (3.06 cm3, 16.58 mmol) was slowly added to a stirred solution of 2,3-O-isopropylidene-D-ribono-1,4-lactone 14 [5] (1.04 g, 5.53 mmol) in DCM (10 cm3) at ambient temperature. After stirring at 40 °C for 30 min, the reaction mixture was cooled down to ambient temperature and silica gel (0.5 g) was carefully added. After removal of solvent, silica gel was applied to a silica gel column eluting with EtOAc:hexane (1:3) to afford 15 as a white solid (0.88 g, 84%); mp: 60–62 °C; [α]20D: −82.8° (c 5.0 × 10−3, CHCl3); 1H NMR (400 MHz; CDCl3): δ = 1.39 (s, 3H, CH3), 1.48 (s, 3H, CH3), 4.67 (ddd, 1H, 3JHH = 1.9, 2JHH = 11.0, 2JHF = 46.1, H-5), 4.72 (dddd, 1H, 3JHH = 1.7, 3JHH = 1.7, 3JHH = 1.7, 3JHF = 34.57, H-4), 4.74 (ddd, 1H, 3JHH = 1.8, 2JHH = 11.0, 2JHF = 48.1, H-5), 4.81 (dd, 1H, 3JHH = 3.3, 3JHH = 5.6, H-3), 4.85 (d, 1H, 3JHH = 5.7, H-2). 13C NMR (100 MHz; CDCl3): δ = 25.5 (CH3), 26.8 (CH3), 75.1 (d, 4JCF = 3.9, C-2), 77.2 (d, 3JCF = 5.1, C-3), 80.3 (d, 2JCF = 19.2, C-4), 82.2 (d, 1JCF = 171.3, C-5), 113.9 (C(CH3)2), 173.6 (C-1). 19F NMR (282 MHz, CDCl3): δ = −236.0 (ddd, 3JHF = 3.3, 2JHF = 34.8, 2JHF = 46.3). νmax (KBr): 3563, 2993, 1804, 1455, 1384, 1272, 1054, 849, 610, 513. HRMS (ES+): Calcd. for C8H11FO4Na [M+Na]+: 213.0539, found: 213.0537.
5-Deoxy-5-fluoro-2,3-O-isopropylidene-D-ribofuranose (17). A solution of DIBAL-H (1 M in hexane, 2.20 ml, 2.18 mmol) was carefully added to a solution of 5-deoxy-5-fluoro-2,3-O-isopropylidene-D-ribono-1,4-lactone (15) (0.42 g, 2.18 mmol) in dry toluene (10 ml) at 40 °C and the reaction was monitored by 19F NMR. After completion, MeOH (2 ml) was added. The slurry gel was broken by adding sat. aq. solution of sodium potassium tartrate (1 ml). After extracted into ether (3 × 10 ml), the combined organic phases were washed with brine and dried over MgSO4. After removal of solvent under reduced pressure the residue was purified over silica gel eluting with 60% EtOAc/hexane to afford 17 as colourless oil (0.26 g, α/β = 7.6/1, 63.6%) and 16 (25.4 mg, 6.0%). α-anomer: 1H NMR (300 MHz; CDCl3): δ = 1.27 (s, CH3), 1.43 (s, CH3), 3.27 (t, 3JHH = 5.17, -OH), 4.31–4.41 (m, 2H, H-4, H-5, (overlap with β, H-4, H-5)), 4.44–4.61 (m, 2H, H-3, H-5 (overlap with β, H-3, H-5)), 4.71 (d, 3JHH = 5.9, H-2), 5.38 (dd, 1H, 3JHH = 2.5, 3JHH = 5.3, H-1). 13C NMR (75 MHz; CDCl3): δ = 24.9 (CH3), 26.5 (CH3), 81.2 (d, 3JCF = 5.5, C-4), 82.2 (d, 1J = 171.3, C-5), 84.8 (C-3 or C-2), 86.1 (C-2 or C-3), 103.3 (C-1), 112.7 (C(CH3)2). 19F NMR (282 MHz, CDCl3): δ = −230.8 (dddd, 4JHF = 3.3, 3JHF = 37.5, 2JHF = 47.3, 2JHF = 47.3, 1F). β-anomer: 1H NMR (300 MHz; CDCl3): δ = 1.50 (s, CH3), 1.33 (s, CH3), 3.93 (d, 3JHH = 10.9, -OH), 4.31–4.41 (m, 2H, H-4, H-5, (overlap with α, H-4, H-5)), 4.44–4.61 (m, 2H, H-3, H-5 (overlap with α, H-3, H-5)), 4.74 (ddd, 1H, 3JHH = 0.8, 3JHH = 1.5, 3JHH = 6.4, H-4), 5.33 (ddd, 1H, 3JHH = 3.7, 3JHH = 3.7, 3JHH = 10.9, H-1). 13C NMR (75 MHz; CDCl3): δ = 24.7 (CH3), 26.1 (CH3), 79.5 (C-2), 79.6 (d, 2JCF = 17.8, C-4), 80.7 (d, 3JCF = 6.7, C-3), 85.1 (d, 1J = 170.5, C-5), 97.7 (C-1), 114.2 (C(CH3)2). 19F NMR (282 MHz, CDCl3): δ = −225.4 (dddd, 4JHF = 3.2, 3JHF = 23.1, 2JHF = 46.5, 2JHF = 46.5, 1F). νmax (neat): 3426, 2944, 1376, 1211, 1072, 868, 504. HRMS (ES+): Calcd. for C8H13FO4Na [M+Na]+: 215.0696, found: 215.0691.
D-allo,altro-2,5-Anhydro-1-deoxy-1-(dimethoxyphosphinyl)-6-fluoro-3,4-O-isopropylidenehexitol (18). 50% aq Solution NaOH (3 ml) was slowly added to a vigorously stirring mixture of 5-deoxy-5-fluoro-2,3-O-isopropylidene-D-ribofuranose 17 (0.125 g, 0.63 mmol) and tetramethyl methylenediphosphonate (0.16 g, 0.69 mmol) in DCM (3 ml) at ambient temperature. After 18 h, the reaction mixture was diluted with DCM (10 ml) and the aq phase was extracted with DCM (2 × 10 ml). The combined organic phases were dried over MgSO4, and filtered. After removal of solvent, the residue was purified over silica gel eluting with 5% MeOH:DCM to afford a mixture of epimers 18 as colourless oil (0.11 g, α/β = 3.2/1, 58.5%).1H NMR (400 MHz; CDCl3): δ = 1.19 (C(CH3)2), 1.33 (C(CH3)2), 1.95–2.20 (m, 2H, H-1), 3.58–3.63 (m, POCH3), 3.95–4.69 (m, 6H). 13C NMR (162 MHz; CDCl3): δ = 30.3–31.1 (m, Pα), 31.7–32.4 (m, Pβ). 19F NMR (282 MHz, CDCl3): δ = −231.1 (ddd, 3J = 28.90, 2J = 47.32, 2J = 47.32, 1Fα), −229.3 (dddd, 4J = 2.26, 3J = 32.94, 2J = 47.29, 2J = 47.29, 1Fβ). νmax (neat): 3466, 2955, 1644, 1459, 1374, 1213, 1023, 822, 537. HRMS (ES+): Calcd. for C11H20O4FPNa [M+Na]+: 321.0879, found: 321.0874.
D-altro-2,5-Anhydro-1-deoxy-1-(dimethoxyphosphinyl)-6-fluoro-3,4-O-isopropylidenehexitol (23a). A solution of TBAF (1 M in THF, 2.16 ml, 2.16 mmol) was added to a mixture of 22a (0.16 g, 0.54 mmol) and TsF (0.28g, 1.62 mmol) in THF (15 ml) and then the reaction mixture was brought to reflux. After refluxing for 18 h, the reaction mixture was cooled down to ambient temperature and then filtered through a shot pad of silica gel washing with EtOAc. After removal of solvent the residue was purified over silica gel eluting with MeOH:DCM (1:20) to afford 23a as colourless oil (0.148 g, 92.3%). [α]D20: −16.44° (c 2.25 × 10−3, CHCl3). 1H NMR (300 MHz; CDCl3): δ = 1.28 (s, 3H, C(CH3)2). 1.42 (s, 3H, C(CH3)2), 2.13 (ddd, 1H, 3JHH = 6.7, 2JHH = 15.4, 2JHP = 18.1, H-1), 2.23 (ddd, 1H, 3JHH = 6.6, 2JHH = 15.4, 2JHP = 18.4, H-1), 3.68 (d, 3H, 3JHH = 3.9, POCH3), 3.70 (d, 3H, 3JHH = 3.9, POCH3), 4.12 (ddd, 1H, 3JHH = 2.9, 3JHH = 2.9, 3JHF = 33.1, H-5), 4.26 (dddd, 1H, 3JHH = 3.2, 3JHH = 6.6, 3JHH = 6.6, 3JHP = 10.3, H-2), 4.44 (ddd, 1H, 3JHH = 3.4, 2JHH = 10.2, 2JHF = 46.8, H-5), 4.48 (ddd, 1H, 3JHH = 3.2, 2JHH = 10.2, 2JHF = 47.6, H-5), 4.65 (dd, 3JHH = 3.9, 3JHH = 6.0, 1H, H-3), 4.77 (d, 3JHH = 6.0, 1H, H-4). 13C NMR (75 MHz; CDCl3): δ = 24.9 (CH3), 25.5 (d, 2JPC = 141.9, C-1), 26.2 (CH3), 52.3 (d, 2JPC = 6.4, POCH3), 52.6 (d, 2JPC = 6.2, POCH3), 76.9 (d, 2JCF = 1.9, C-2), 81.7 (d, 2JPC = 7.5, C-3), 82.1 (d, 2JCF = 6.4, C-4), 82.5 (d, 2JCF = 17.8, C-5), 84.8 (d, 1JCF = 172.4, C-6), 112.7 (C(CH3)2). 31P NMR (121 MHz; CDCl3): δ = 31.65–32.38 (m, P). 19F NMR (282 MHz; CDCl3) −229.3 (ddd, 3JHF = 2.2, 3JHF = 33.2, 2JHF = 33.2, 2JHF = 47.1, 1F). νmax (neat): 2954, 1372, 1209, 1025, 819, 503. HRMS (ES+): Calcd. for C11H20O4FPNa [M+Na]+: 321.0879, found: 321.0879.
D-allo-2,5-Anhydro-1-deoxy-1-(dimethoxyphosphinyl)-6-fluoro-3,4-O-isopropylidenehexitol (23b). The same procedure as described for 23a was repeated with 22b (70 mg, 0.23 mmol) to afford 23b as a colourless oil (60 mg, 83.1%). [α]D20: −18.78° (c 2.45 × 10−3, CHCl3), 1H NMR (300 MHz; CDCl3): δ = 4.64 (dd, 3JHH = 3.9, 3JHH = 6.6, 1H, H-4), 4.49 (ddd, 1H, 3JHH = 2.8, 2JHH = 10.4, 2JHF = 47.6, H-6), 4.48 (dd, 1H, 3JHH = 4.3, 3JHH = 6.6, H-3), 4.45 (ddd, 1H, 3JHH = 3.5, 2JHH = 10.8, 2JHF = 47.0, H-6), 4.23 (dddd, 1H, 3JHH = 4.2, 3JHH = 6.7, 3JHH = 6.7, 3JHP = 11.1, H-2), 4.09 (ddd, 1H, 3JHH = 3.4, 3JHH = 3.4, 2JHF = 28.3, H-5), 3.71 (d, 3H, 3JHH = 2.2, POCH3), 3.67 (d, 3H, 3JHH = 2.3, POCH3), 2.09 (d, 1H, 2JHP = 18.4, H-1), 2.13 (dd, 1H, 3JHH = 0.9, 2JHP = 18.4, H-1), 1.47 (s, 3H, C(CH3)2), 1.28 (s, 3H, C(CH3)2). 13C NMR (75 MHz; CDCl3): δ = 25.9 (CH3), 27.7 (CH3), 30.1 (d, 2JCP = 141.1, C-1), 52.7 (d, 2JCP = 6.5, OCH3), 52.9 (d, 2JCP = 6.5, OCH3), 80.5 (d, 2JCF = 3.4, C-2), 81.2 (d, 3JCF = 7.4, C-4), 83.2 (d, 2JCF = 172.7, C-6), 83.5 (d, 2JCF = 18.3, C-5), 85.7 (d, 4JCF = 11.3, C-3), 114.9 (C(CH3)2). 31P NMR (162 MHz; CDCl3): δ = 30.33−31.09 (m, P). 19F NMR (282 MHz; CDCl3): δ = −231.0 (ddd, 3JHF = 28.8, 2JHF = 47.3, 2JHF = 47.3, 1F). νmax (neat): 3464, 2956, 1383, 1214, 1030, 822, 513. HRMS (ES+): Calcd. for C11H20O4FPNa [M+Na]+: 321.0879, found: 321.0875.
Cyclohexylammonium salt 8a. TMSBr (0.18 ml, 1.36 mmol) was added to a solution of 23a (94.8 mg, 0.34 mmol) in DCM (5 ml). The progress of reaction was monitored by 31P NMR. The disappearing of signal at 31.5–32.1.0 ppm indicating that the reaction was gone completion. A solution of TFA:H2O (1:1, 1 ml) was then added to the mixture. After 30 min, water (5 ml) was added, the solvents were removed under reduced pressure. To this residue was added water (5 ml) and the solvent was removed under reduced pressure; this was repeated until all trace of TFA was gone. To this residue 5 ml of water was added and adjusted to pH 11 with cyclohexylamine. After removal of solvent, the residue was dissolved in EtOH and about 0.5 volume of acetone was added. The precipitate was filtered off to afford 8a as colourless needles (97.6 mg, 87.2%). Mp. 168–170 °C, [α]D20: +18.67° (c 1.50 × 10−3, MeOH), 1H NMR (400 MHz; MeOH-d4): δ = 1.07–1.16 (m, 1H, cyclohexyl), 1.19–1.34 (m, 4H, cyclohexyl), 1.57–1.64 (m, 1H, cyclohexyl), 1.70–1.79 (m, 2H, cyclohexyl), 1.88–1.94 (m, 2H, cyclohexyl), 1.87 (ddd, 1H, 3JHH = 4.9, 2JHH = 10.1, 2JHP = 19.1, H-1), 2.04 (ddd, 1H, 3JHH = 10.1, 2JHH = 14.2, 2JHP = 17.1, H-1), 2.91–2.98 (m, 1H, H-1, cyclohexyl), 3.83 (dddd, 1H, 3JHH = 2.0, 3JHH = 4.6, 3JHH = 8.7, 3JHF = 25.5, H-5), 4.01 (dd, 1H, 3JHH = 4.6, 3JHH = 8.7, H-4), 4.10–4.14 (br, 1H, H-3), 4.17 (dddd, 1H, 3JHH = 2.9, 3JHH = 4.5, 3JHH = 4.5, 3JHP = 9.7, H-2), 4.34 (ddd, 1H, 3JHH = 4.7, 2JHH = 10.3, 2JHF = 47.7, H-6), 4.56 (ddd, 1H, 3JHH = 2.1, 2JHH = 10.3, 2JHF = 48.4, H-6). 13C NMR (100 MHz; MeOH-d4): δ = 25.4 (2 × C, cyclohexyl), 25.9 (C, cyclohexyl), 31.2 (d, 1JCP = 129.5, C-1), 31.9 (2 × C, cyclohexyl), 51.5 (C-1, cyclohexyl), 72.9 (d, 3JCF = 7.9, C-4), 73.4 (C-3), 79.7 (d, 2JCP = 2.7, C-2), 81.0 (d, 2JCF = 17.6, C-5), 84.3 (d, 1JCF = 170.9, C-6). 31P NMR (162 MHz; MeOH-d4): δ = 18.7 (ddd, 3JHP = 2.9, 2JHP = 18.1, 2JHP = 18.1 ). 19F NMR (376 MHz; MeOH-d4): δ = −232.4 (ddd, 3JHF = 25.5, 2JHF = 47.4, 2JHF = 47.4, 1F). νmax (KBr): 3199, 2939, 2720, 2012, 1597, 1429, 1389, 1121, 1018, 925. HRMS (ES+): Calcd. for C12H26NO6FP [M+H]+: 330.1482, found: 330.1492.
Cyclohexylammonium salt 8b. The same procedure as described for preparation of 8a was repeated with 23b (53.1 mg, 0.19 mmol) to afford 8b as white solid (52.1 mg, 83.3%). Mp. 146–148 °C, [α]D20: +3.09° (c 3.55 × 10−3, MeOH), 1H NMR (400 MHz; MeOH-d4): δ = 1.05–1.32 (m, 5H, cyclohexyl), 1.54–1.74 (m, 5H, 1H-1 overlap with 4H-cyclohexyl), 1.87–1.90 (m, 2H, cyclohexyl), 1.99 (ddd, 1H, 3JHH = 3.7, 2JHH = 14.1, 2JHP = 18.8, H-1), 2.83–2.91 (m, 1H, H-1, cyclohexyl), 3.96 (t, 3JHH = 6.3, H-3), 3.83 (dddd, 1H, 3JHH = 3.7, 3JHH = 3.7, 3JHH = 3.7, 3JHF = 25.9, H-5), 3.97 (dd, 1H, 3JHH = 3.9, 3JHH = 5.5, H-4), 3.95–4.01 (m, 1H, H-2), 4.31 (ddd, 1H, 3JHH = 4.1, 2JHH = 10.2, 2JHF = 47.4, H-6), 4.36 (ddd, 1H, 3JHH = 3.2, 2JHH = 10.2, 2JHF = 47.9, H-6). 13C NMR (100 MHz; MeOH-d4): δ = 25.5 (2 × C, clohexyl), 26.1 (C, cyclohexyl), 32.8 (2 × C, clohexyl), 35.9 (d, 1JCP = 127.0, C-1), 51.4 (C-1, cyclohexyl), 73.1 (d, 3JCF = 5.1, C-4), 77.4 (C-3), 80.7 (d, 2JCP = 2.6, C-2), 84.1 (d, 2JCF = 18.5, C-5), 84.3 (d, 1JCF = 170.5, C-6). 31P NMR (162 MHz; MeOH-d4): δ = 17.9 (ddd, 3JHP = 3.9, 2JHP = 17.8, 2JHP = 17.8 ). 19F NMR (376 MHz; MeOH-d4): δ = −231.6 (ddd, 1F, 3JHF = 25.6, 2JHF = 47.7, 2JHF = 47.7). νmax (KBr): 3436, 2937, 2561, 2012, 1618, 1454, 1389, 1051, 974. HRMS (ES+): Calcd. for C12H26NO6FP [M+H]+: 330.1482, found: 330.1485.
Fluoromethylphostone 10
Deprotection as described for 8a was repeated with 23a (80 mg, 0.27 mmol) in DCM (5 ml). Without addition of cyclohexylamine, the residue was dissolved in dry pyridine (1 ml) and acetic anhydride (0.04 ml, 0.4 mmol) was then added. The emergence of an 31P-NMR signal at 45 ppm and a disappearance of signal at 27 ppm indicated that the reaction had gone completion. After removal of the solvent under reduced pressure, the crude was purified over silica gel eluting with MeOH:DCM (3:2) to afford 10 as white solid (23 mg, 40.2%). Mp. 180−182 °C, [α]D20: +103.0° (c 1.0 × 10−3, MeOH). 1H NMR (300 MHz; MeOH-d4): δ = 1.73–1.98 (m, 2H, H-1), 4.25 (dddd, 1H, 3JHH = 2.10, 3JHH = 3.90, 3JHH = 8.30, 3JHF = 26.32, H-1, H-5), 4.36 (ddd, 1H, 3JHH = 3.90, 2JHH = 10.63, 2JHF = 47.18, H-6), 4.48 (ddd, 1H, 3JHH = 2.10, 2JHH = 10.63, 2JHF = 48.14, H-6), 4.67–4.71 (m, 1H, H-3), 4.72–4.82 (m, 1H, H-2), 4.88 (ddd, 1H, 3JHH = 1.91, 3JHH = 4.61, 3JHP = 8.34, H-4). 13C NMR (75 MHz; MeOH-d4): δ = 29.6 (d, 1JCP = 119.5, C-1), 74.4 (d, 4JCF = 7.15, C-4), 77.8 (d, 4JCF = 8.8, C-3), 79.5 (d, 3JCF = 18.36, C-5), 81.2 (d, 2JCP = 4.0, C-2), 83.2 (d, 1JCF = 172.1, C-6). 19F NMR (282 MHz; MeOH-d4): δ = −234.3 (ddd, 1F, 3JHF = 26.4, 2JHF = 47.4, 2JHF = 47.7). 31P NMR (162 MHz; MeOH-d4): δ = 41.9–42.6 (br, m). νmax (PTFE): 3419, 1214, 1154, 799, 503. HRMS (ES-): Calcd. for C6H9FO5P [M-H]: 211.0172, found: 211.0166.
Supporting Information
The experimental and analytical data for compounds 9b, 11, 14, 16, and 19–22 are provided in the supporting information.
| Supporting Information File 1: Experimental. | ||
| Format: DOC | Size: 87.5 KB | Download |
References
-
Sanada, M.; Miyano, T.; Iwadare, S.; Williamson, J. M.; Arison, B. H.; Smith, J. L.; Douglas, A. W.; Liesch, J. M.; Inamine, E. J. Antibiot. 1986, 39, 259–265.
Return to citation in text: [1] -
Deng, H.; Cross, S. M.; McGlinchey, R. P.; Hamilton, J.; O'Hagan, D. Chem. Biol. 2008, 15, 1268–1276. doi:10.1016/j.chembiol.2008.10.012
Return to citation in text: [1] [2] -
O'Hagan, D.; Schaffrath, C.; Cobb, S. L.; Hamilton, J. T. G.; Murphy, C. D. Nature 2002, 416, 279. doi:10.1038/416279a
Return to citation in text: [1] -
Meyer, R. L.; Stone, T. E.; Jesthi, P. K. J. Med. Chem. 1984, 27, 1095–1098. doi:10.1021/jm00374a028
Return to citation in text: [1] [2] [3] -
Kandil, A. A.; Slessor, K. N. J. Org. Chem. 1985, 50, 5649–5655. doi:10.1021/jo00350a045
Return to citation in text: [1] [2] -
Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M. Chem. Commun. 1999, 215–216. doi:10.1039/a808517j
Return to citation in text: [1] -
Fox, D. T.; Poulter, C. D. J. Org. Chem. 2005, 70, 1978–1985. doi:10.1021/jo048022h
Return to citation in text: [1] -
Baker, S. J.; Young, D. W. J. Labelled Compd. Radiopharm. 2000, 43, 1023–1032. doi:10.1002/1099-1344(200009)43:10<1023::AID-JLCR388>3.0.CO;2-B
Return to citation in text: [1] -
Kohli, V.; Blöcker, H.; Köster, H. Tetrahedron Lett. 1980, 21, 2683–2686. doi:10.1016/S0040-4039(00)78579-0
Return to citation in text: [1] -
Shimuzu, M.; Nakahara, Y.; Yoshioka, H. Tetrahedron Lett. 1985, 26, 4207–4210. doi:10.1016/S0040-4039(00)98993-7
Return to citation in text: [1] -
Ashton, T. D.; Scammells, P. Bioorg. Med. Chem. Lett. 2005, 15, 3361–3363. doi:10.1016/j.bmcl.2005.05.029
Return to citation in text: [1] -
McClard, r. W.; Witte, J. F. Bioorg. Med. Chem. Lett. 1994, 4, 1537–1538. doi:10.1016/S0960-894X(01)80528-X
Return to citation in text: [1] -
Bosco, M.; Bisseret, P.; Eustache, J. Tetrahedron Lett. 2003, 44, 2347–2349. doi:10.1016/S0040-4039(03)00240-5
Return to citation in text: [1]
| 1. | Sanada, M.; Miyano, T.; Iwadare, S.; Williamson, J. M.; Arison, B. H.; Smith, J. L.; Douglas, A. W.; Liesch, J. M.; Inamine, E. J. Antibiot. 1986, 39, 259–265. |
| 4. | Meyer, R. L.; Stone, T. E.; Jesthi, P. K. J. Med. Chem. 1984, 27, 1095–1098. doi:10.1021/jm00374a028 |
| 13. | Bosco, M.; Bisseret, P.; Eustache, J. Tetrahedron Lett. 2003, 44, 2347–2349. doi:10.1016/S0040-4039(03)00240-5 |
| 3. | O'Hagan, D.; Schaffrath, C.; Cobb, S. L.; Hamilton, J. T. G.; Murphy, C. D. Nature 2002, 416, 279. doi:10.1038/416279a |
| 5. | Kandil, A. A.; Slessor, K. N. J. Org. Chem. 1985, 50, 5649–5655. doi:10.1021/jo00350a045 |
| 2. | Deng, H.; Cross, S. M.; McGlinchey, R. P.; Hamilton, J.; O'Hagan, D. Chem. Biol. 2008, 15, 1268–1276. doi:10.1016/j.chembiol.2008.10.012 |
| 10. | Shimuzu, M.; Nakahara, Y.; Yoshioka, H. Tetrahedron Lett. 1985, 26, 4207–4210. doi:10.1016/S0040-4039(00)98993-7 |
| 11. | Ashton, T. D.; Scammells, P. Bioorg. Med. Chem. Lett. 2005, 15, 3361–3363. doi:10.1016/j.bmcl.2005.05.029 |
| 2. | Deng, H.; Cross, S. M.; McGlinchey, R. P.; Hamilton, J.; O'Hagan, D. Chem. Biol. 2008, 15, 1268–1276. doi:10.1016/j.chembiol.2008.10.012 |
| 12. | McClard, r. W.; Witte, J. F. Bioorg. Med. Chem. Lett. 1994, 4, 1537–1538. doi:10.1016/S0960-894X(01)80528-X |
| 8. | Baker, S. J.; Young, D. W. J. Labelled Compd. Radiopharm. 2000, 43, 1023–1032. doi:10.1002/1099-1344(200009)43:10<1023::AID-JLCR388>3.0.CO;2-B |
| 4. | Meyer, R. L.; Stone, T. E.; Jesthi, P. K. J. Med. Chem. 1984, 27, 1095–1098. doi:10.1021/jm00374a028 |
| 7. | Fox, D. T.; Poulter, C. D. J. Org. Chem. 2005, 70, 1978–1985. doi:10.1021/jo048022h |
| 9. | Kohli, V.; Blöcker, H.; Köster, H. Tetrahedron Lett. 1980, 21, 2683–2686. doi:10.1016/S0040-4039(00)78579-0 |
| 6. | Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M. Chem. Commun. 1999, 215–216. doi:10.1039/a808517j |
| 5. | Kandil, A. A.; Slessor, K. N. J. Org. Chem. 1985, 50, 5649–5655. doi:10.1021/jo00350a045 |
| 4. | Meyer, R. L.; Stone, T. E.; Jesthi, P. K. J. Med. Chem. 1984, 27, 1095–1098. doi:10.1021/jm00374a028 |
© 2009 Nasomjai et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)