Search results
Search for "molecular dynamics" in Full Text gives 109 result(s) in Beilstein Journal of Organic Chemistry.
Peptide stapling by late-stage Suzuki–Miyaura cross-coupling
Beilstein J. Org. Chem. 2022, 18, 1–12, doi:10.3762/bjoc.18.1

- its native binding partner β-catenin. An increased proteolytic stability against proteinase K has been demonstrated. Keywords: accelerated molecular dynamics; halotryptophan; intrinsically disordered peptides; late-stage diversification; macrocyclisation; molecular dynamics; stapled peptides; Suzuki
- cyclised RGD peptides. It could be proven that an isomerisation is not caused by the cross-coupling but by the presence of stable isomers/conformers. Molecular dynamics (MD) simulations verified the appearance of stable, distinct conformers or atropisomers, which were in accordance with the experimental
- . The conformational preferences of the stapled peptide P5 and of the linear peptides P6 and aAxWt were investigated via extensive accelerated molecular dynamics simulations (aMD) as implemented within the Amber18 program package [85]. The aMD methodology developed by McCammon and co-workers [86] has








GlycoBioinformatics
Beilstein J. Org. Chem. 2021, 17, 2726–2728, doi:10.3762/bjoc.17.184

- article by Barnett et al. [2] uses molecular dynamics to show that O-linked glycosylation alters peptide conformation, which influences the binding of the peptides to antibodies, despite the fact that glycans are not directly involved in the binding. Another molecular modeling article by Fogarty et al. [3
- one of the authors of this article, Fadda, used glyco-adapted molecular dynamics to explain in a separate publication [4] how the COVID-19 spike protein recognition element requires N-linked glycosylation to be exposed. Another approach to understanding glyco-interactions is described in a review
(Phenylamino)pyrimidine-1,2,3-triazole derivatives as analogs of imatinib: searching for novel compounds against chronic myeloid leukemia
Beilstein J. Org. Chem. 2021, 17, 2260–2269, doi:10.3762/bjoc.17.144

- docking results were edited using the Visual Molecular Dynamics 1.9.3 (VMD) program (available for download at http://www.ks.uiuc.edu/Research/vmd/vmd-1.9.3/). Proposed structural modifications to obtain triazole derivatives 1a, b and 2a–j. Asymmetric unit representation of the 1,2,3-triazole derivative






Double-headed nucleosides: Synthesis and applications
Beilstein J. Org. Chem. 2021, 17, 1392–1439, doi:10.3762/bjoc.17.98

- . The analysis of the melting temperature of the duplex and extensive molecular dynamics studies revealed that the synthesized double-headed nucleotides behave as functional dinucleotide mimics and hybridize with complementary targets neatly with their Watson–Crick faces compatible with natural DNA [39
- that resulted in the formation of two novel nucleic acid motifs. The novel nucleic acid motifs could be incorporated either single or multiple times in dsDNA duplexes without altering its stability. It was revealed by molecular dynamics (MD) simulations that the DNA sugar–phosphate backbone
- strands. Oligonucleotides with fourteen consecutive incorporations of different double-headed nucleosides were synthesized and the DNA duplexes showed increased stability owing to increased stacking interactions among the nucleobases of the opposite strands [72]. Molecular dynamics simulations







































































Biochemistry of fluoroprolines: the prospect of making fluorine a bioelement
Beilstein J. Org. Chem. 2021, 17, 439–460, doi:10.3762/bjoc.17.40

















19F NMR as a tool in chemical biology
Beilstein J. Org. Chem. 2021, 17, 293–318, doi:10.3762/bjoc.17.28

- distance restraints for ion channels and other protein complexes that would be difficult to be defined by using other analytical tools. DNA and RNA secondary and tertiary structure 19F NMR spectroscopy also represents a useful analytical approach to study the structure, function and molecular dynamics of























Comparative ligand structural analytics illustrated on variably glycosylated MUC1 antigen–antibody binding
Beilstein J. Org. Chem. 2020, 16, 2540–2550, doi:10.3762/bjoc.16.206

- approach by investigating the in-silico binding of a peptide and glycopeptide epitope of the glycoprotein Mucin 1 (MUC1) binding with the antibody AR20.5. To study the binding, we performed molecular dynamics simulations using OpenMM and then used the Galaxy platform for data analysis. The same analysis
- . and others [14][16] provides a foundation for further investigation into the binding of glycopeptide antigens to antibodies using computational modeling. Molecular dynamics (MD) simulations and analysis thereof are a well-known ingredient of the in-silico process for mechanistic screening of
- of the analyses was carried out using Galaxy, the popular open web-based platform for bioinformatics and computational data analysis, which enables the creation of repeatable analysis pipelines (workflows). There are several well-known molecular dynamics analysis packages (MDAnalysis [37], Bio3D [38






Leveraging glycomics data in glycoprotein 3D structure validation with Privateer
Beilstein J. Org. Chem. 2020, 16, 2523–2533, doi:10.3762/bjoc.16.204

- study of these glycan-mediated interactions can provide unique insight into the molecular interplay governing these processes. In addition, it can provide structural snapshots in atomistic detail that can be used to generate molecular dynamics simulations describing a wider picture underpinning glycan
- /glycoproteomics to carbohydrate 3D model building and validation in Privateer Many fields, for example pharmaceutical design and engineering [58], molecular dynamics simulations [59] and protein interaction studies [60], rely upon structural biology to produce accurate atomistic descriptions of glycoproteins





Computational tools for drawing, building and displaying carbohydrates: a visual guide
Beilstein J. Org. Chem. 2020, 16, 2448–2468, doi:10.3762/bjoc.16.199

- , 3D representation of structures is provided by tools such as Visual Molecular Dynamics (VMD) [21] , and LiteMol [22], which allow for quick analysis of structural features in 3D space. All the tools mentioned were evaluated against a set of pre-selected criteria relating to ease of use, scientific
- models. These tools generally use 3D molecular templates of monosaccharides to reconstruct a 3D model. Energy minimisation methods can further refine the models. These models are essential for structure-based studies and complex calculations like Molecular Dynamics simulations. Therefore, the accurate
- incorporating SNFG symbols in 3D space for further visualisation using the computational tools like visual molecular dynamics (VMD) [21] LiteMol [22] and Sweet Unity Mol [32]. Glycan sketchers SugarSketcher. SugarSketcher [14] is a JavaScript interface module currently included in the tool collection of












How and why plants and human N-glycans are different: Insight from molecular dynamics into the “glycoblocks” architecture of complex carbohydrates
Beilstein J. Org. Chem. 2020, 16, 2046–2056, doi:10.3762/bjoc.16.171

- undetected because of their intrinsic structural disorder. In this work we use molecular dynamics (MD) simulations to provide insight into N-glycans’ 3D structure by analysing the effects of a set of very specific modifications found in plants and invertebrate N-glycans, which are immunogenic in humans. We
- relationships in complex carbohydrates, with important implications in glycoengineering design. Keywords: complex carbohydrates; fucose; glycoblocks; molecular dynamics; molecular recognition; N-glycans; xylose; Introduction Complex carbohydrates (or glycans) are an essential class of biomolecules, directly
- glycans sequence can alter their 3D structure and conformational dynamics, ultimately regulating recognition [19]. In this work we use molecular dynamics (MD) simulations to analyse the effects of the inclusion of motifs typically found in plants and invertebrates N-glycans and immunogenic in mammals [20








pH- and concentration-dependent supramolecular self-assembly of a naturally occurring octapeptide
Beilstein J. Org. Chem. 2020, 16, 2017–2025, doi:10.3762/bjoc.16.168

- (Glu−) amino acids [73][74]. Mechanistic insights into the observed conformational transformation via molecular dynamics simulations are underway in our laboratory. Conclusion In summary, we synthesized a naturally occurring amphiphilic peptide fragment, PEP-1, from a β-sheet lectin protein, galectin-1






Synthesis, docking study and biological evaluation of ᴅ-fructofuranosyl and ᴅ-tagatofuranosyl sulfones as potential inhibitors of the mycobacterial galactan synthesis targeting the galactofuranosyltransferase GlfT2
Beilstein J. Org. Chem. 2020, 16, 1853–1862, doi:10.3762/bjoc.16.152

- mechanism studies using computational chemistry methods. The probable reaction mechanisms were studied by hybrid DFT QM/MM molecular dynamics simulations [11] where the possible transition state (TS) structures were localized. The observation of the possible TS structure opens the opportunities for the in
- structures. All fructofuranose and tagatofuranose derivatives were docked into the GlfT2 structure obtained by QM/MM molecular dynamics simulations, which representes the structure close to the transition state structure of the GlfT2 catalytic reaction. The best ten docking poses of each docked molecule were










Models of necessity
Beilstein J. Org. Chem. 2020, 16, 1649–1661, doi:10.3762/bjoc.16.137

- become apparent [40]. This is because non-equilibrium structures such as transition states that cannot be described by the conventional Lewis model are passed through during reactions. Quantum chemical methods, often in combination with molecular dynamics, can predict the course of a specific reaction





Highly selective Diels–Alder and Heck arylation reactions in a divergent synthesis of isoindolo- and pyrrolo-fused polycyclic indoles from 2-formylpyrrole
Beilstein J. Org. Chem. 2020, 16, 1320–1334, doi:10.3762/bjoc.16.113

- stereoselective approaches of substrates and reagents or catalysts in a variety of processes. Examples include the substrate–enzyme recognition (the host–guest interaction) responsible for inducing pharmacological activity, the DNA-intercalation capable of generating biomolecular activity, molecular dynamics [63













Towards the total synthesis of chondrochloren A: synthesis of the (Z)-enamide fragment
Beilstein J. Org. Chem. 2020, 16, 670–673, doi:10.3762/bjoc.16.64

- of the molecule was elucidated by a combination of NMR, UV and IR spectroscopy and molecular dynamics calculations (MD, MM2) [12]. However, its (Z)-enamide motif and the polyoxygenated middle segment are synthetically challenging. Results and Discussion Synthesis of amide 3 Here we report our





Photocontrolled DNA minor groove interactions of imidazole/pyrrole polyamides
Beilstein J. Org. Chem. 2020, 16, 60–70, doi:10.3762/bjoc.16.8
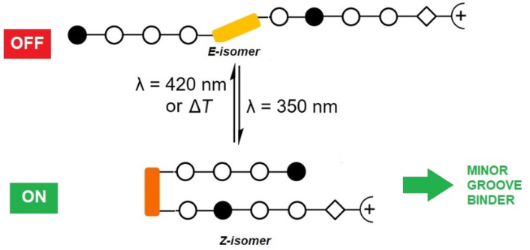
- ]. According to molecular dynamics calculations, the 3,3'-substituted azobenzenes are more suitable as photoswitchable building blocks to induce a hairpin motif than the 4,4'-substituted correlates [35]. For 3,3'-substituted azobenzenes, the Z-form is expected to display higher thermal stability than for the







1,2,3-Triazolium macrocycles in supramolecular chemistry
Beilstein J. Org. Chem. 2019, 15, 2142–2155, doi:10.3762/bjoc.15.211

- concentration (10−5 mol/L) in a competitive aqueous–organic CHCl3/CH3OH/H2O mixture (45:45:10, v/v/v). Fluorescence spectroscopy measurements supported by molecular dynamics simulation data have revealed that the smaller macrocyclic rings move from the core-substituted PDI motif to the two 1,2,3-triazolium

















Archangelolide: A sesquiterpene lactone with immunobiological potential from Laserpitium archangelica
Beilstein J. Org. Chem. 2019, 15, 1933–1944, doi:10.3762/bjoc.15.189
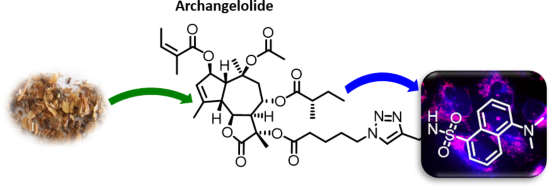
- . Interestingly, we found that neither archangelolide nor its dansyl conjugate did exhibit cytotoxic effects in contrast to the structurally closely related counterparts trilobolide and thapsigargin. We explain this observation by a molecular dynamics simulation, in which, in contrast to trilobolide
- that this SL does not act as SERCA inhibitor. Therefore, we proceeded to confirm this hypothesis by a molecular dynamics simulation study. Molecular dynamics simulation of compounds 1 and 2 with SERCA The binding cavity for thapsigargin and compound 2 in the SERCA protein lies in the transmembrane
- of 10.309 × 10.111 × 15.063 nm3 with periodical boundary conditions and centered. For simulations of energy minimization and molecular dynamics, Gromacs-4.5.5 software was used. The first part of the simulations occurred in vacuum, the other then in water. In order to keep the solution neutral, 24












Water inside β-cyclodextrin cavity: amount, stability and mechanism of binding
Beilstein J. Org. Chem. 2019, 15, 1592–1600, doi:10.3762/bjoc.15.163

- disordered and mobile, and that the OH groups of the host β-CD may rotate [17]. Studies with molecular dynamics simulations have found only four water molecules inside the host β-CD cavity [18]. The energetics of the CD hydration/dehydration have been investigated as well. Experimental studies have been able
- details Different molecular modeling methods (quantum mechanics (QM), molecular dynamics (MD), docking and quantitative structure activity relationships (QSARs)) can be applied in studying the structure, dynamics, and energetics of the host CD systems. However, the results from different modeling (or







Synthesis and conformational preferences of short analogues of antifreeze glycopeptides (AFGP)
Beilstein J. Org. Chem. 2019, 15, 1581–1591, doi:10.3762/bjoc.15.162

- into the ice lattice [12], others investigated the adsorption process on different surfaces via atomic force microscopy [10][13][14]. The antifreeze activity has been correlated with long-range perturbation of hydration dynamics [15]. Latest molecular dynamics simulations suggest that AFGP reversibly






Transient and intermediate carbocations in ruthenium tetroxide oxidation of saturated rings
Beilstein J. Org. Chem. 2019, 15, 1552–1562, doi:10.3762/bjoc.15.158

- can lead to different products in a ratio that depends on reaction dynamics [31][32][33]. The study of molecular dynamics trajectories has allowed characterization of ambimodal transition states in reactions involving carbocations [34][35]. We have demonstrated computationally the presence of












Synthesis and biological evaluation of truncated derivatives of abyssomicin C as antibacterial agents
Beilstein J. Org. Chem. 2019, 15, 1468–1474, doi:10.3762/bjoc.15.147

- docking of known and proposed ligands. The crystal structure contains a tryptophan molecule in the active site. Restrained molecular dynamics [20][21] was employed to position the active site cysteine (Cys-263) in a position that would allow covalent binding of the ligands in the active site. The







Host–guest interactions between p-sulfonatocalix[4]arene and p-sulfonatothiacalix[4]arene and group IA, IIA and f-block metal cations: a DFT/SMD study
Beilstein J. Org. Chem. 2019, 15, 1321–1330, doi:10.3762/bjoc.15.131

- anchoring points for the positively charged guests. Cation–π interactions between the monoatomic cations and p-sulfonatocalix[4]arene in water are supposed (but not proven) to take part in the inclusion complex formation [31]. Mendes et al. have carried out molecular dynamics (MD) simulations of association








Stereo- and regioselective hydroboration of 1-exo-methylene pyranoses: discovery of aryltriazolylmethyl C-galactopyranosides as selective galectin-1 inhibitors
Beilstein J. Org. Chem. 2019, 15, 1046–1060, doi:10.3762/bjoc.15.102





Adhesion, forces and the stability of interfaces
Beilstein J. Org. Chem. 2019, 15, 106–129, doi:10.3762/bjoc.15.12










