Search results
Search for "calculations" in Full Text gives 760 result(s) in Beilstein Journal of Organic Chemistry. Showing first 200.
Heterogeneous photocatalysis in flow chemical reactors
Beilstein J. Org. Chem. 2020, 16, 1495–1549, doi:10.3762/bjoc.16.125

- measurements, TRPL spectroscopy, and density functional theory (DFT) calculations. They found that the HER efficiency correlated to the excited state lifetime and exciton binding energy. The FSO-BP and FSO-FSz hindered the charge transfer and mobility due to the phenyl–phenyl dihedral angle or sharp bends in

















































In silico rationalisation of selectivity and reactivity in Pd-catalysed C–H activation reactions
Beilstein J. Org. Chem. 2020, 16, 1465–1475, doi:10.3762/bjoc.16.122
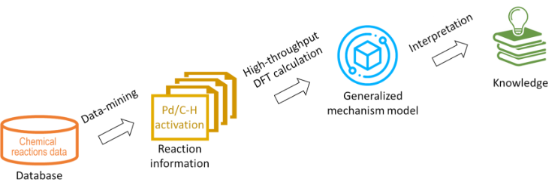
- -driven statistical methods. Here we demonstrate an approach that was developed to automate the DFT-level calculations of energies of the auto-generated reaction intermediates. These results were further used to generalize mechanistic knowledge of a class of reactions, and the developed models were used
- computational data, a threshold to distinguish between two possible reaction mechanisms was established. Computational Methods The NWChem, an open source software package, was used for the DFT calculations. It is easily scalable and designed to solve large scientific computational problems efficiently employing
- , reaction conditions, and so on [23]. In order to achieve accurate and efficient reaction prediction, a mechanism-based method was chosen to direct quantum chemistry calculations and predictions, see Figure 1. For the Pd(II)-catalysed C–H activation reactions, there are two main commonly accepted mechanisms



Recent synthesis of thietanes
Beilstein J. Org. Chem. 2020, 16, 1357–1410, doi:10.3762/bjoc.16.116

- diradicals as intermediates. To explain the regioselectivity, theoretical calculations were performed with thiochalcone and acrylonitrile as model substrates. For the frontier molecular orbital treatment, the largest coefficients in both HOMO and LUMO of thiochalcone existed on the sulfur atom, while the












































































































































Highly selective Diels–Alder and Heck arylation reactions in a divergent synthesis of isoindolo- and pyrrolo-fused polycyclic indoles from 2-formylpyrrole
Beilstein J. Org. Chem. 2020, 16, 1320–1334, doi:10.3762/bjoc.16.113

- ]. Theoretical calculations The Diels–Alder reaction of vinylpyrroles 8b, 8c and 8g with maleimides 7b and 7c resulted in a highly diastereoselective cycloaddition leading to the mixture of endo/exo cycloadducts 9/10, where the endo product 9 was the major one (Scheme 3 and Table 2). Considering the synthetic
- interaction (2.5–3.4 Å and 2.79 Å) [58][59][62][66] according to the calculations and the X-ray calculated average taken from the Cambridge Structural Database (CSD) [76]. Interestingly, in the case of the reaction between diene 8j and 7c, where the diene does not have an N-benzyl group, the endo TS also
- calculations suggest that the greater stability of the endo TSs in the Diels–Alder cycloadditions is associated with NCIs between the N-substituents of both cycloaddends. These interactions are also involved in the case of pyrrolo[2,1-a]isoindole 18a with dienophile 7c. Fused aza-hetero polycyclic frames and













Oxime radicals: generation, properties and application in organic synthesis
Beilstein J. Org. Chem. 2020, 16, 1234–1276, doi:10.3762/bjoc.16.107

- example, based on N-containing heterocycles (isoxazolones, pyrazolones, pyrazolidin-3,5-diones, and 1,2,3-triazolones [52]), sulfones [65], and phosphonates [54] (Scheme 8). Based on the data of EPR spectroscopy [35][38][49][50][66] and quantum chemical calculations [67], the maximum spin density in
- isomers (E and Z) exist. The isomerization of oxime radicals proceeds much easier than for the corresponding oximes; the observation of individual isomers is generally possible only at low temperatures [68][69] (about 190 K). According to quantum chemical calculations, the oxime radicals have an increased
- (77%). When γ,δ-unsaturated oximes were applied the formation of cyclic nitrones was observed (products 107a,b, 108a,b). In this case, the intermediate oxime radicals reacted as N-centered radicals, which was consistent with the calculations [124]. If the double bond of the starting oxime was









































































Development of fluorinated benzils and bisbenzils as room-temperature phosphorescent molecules
Beilstein J. Org. Chem. 2020, 16, 1154–1162, doi:10.3762/bjoc.16.102

- –405 nm (ε: 180–260 M−1·cm−1). To gain more information about the slight difference between the absorption behaviors of the benzil and bisbenzil derivatives, DFT and time-dependent DFT (TD-DFT) calculations at the CAM-B3LYP/6-31+G(d) level of theory were performed for fluorinated benzil 2a and
- corresponding bis-oxidized bisbenzil derivatives. Based on theoretical calculations, the selective formation of the fluorinated analogues stemmed from the slight modulation of the charge distribution at the alkyne moiety of the reactant induced by the electron-withdrawing fluorine atoms. Evaluation of the
- derivatives previously developed by our group and (B) phosphorescent molecular structures intended for this work. Mulliken charge distributions of fluorinated 1a and nonfluorinated 1c obtained from density functional theory calculations [CAM-B3LYP/6-31+G(d) level]. Absorption and photoluminescence (PL








Synthesis and properties of quinazoline-based versatile exciplex-forming compounds
Beilstein J. Org. Chem. 2020, 16, 1142–1153, doi:10.3762/bjoc.16.101

- compounds, based on theoretical calculations and experimental measurements, as well as the electrochemical and thermal properties, are discussed. The synthesized compounds form glasses with glass-transition temperatures ranging from 116 °C to 123 °C. The ionization potentials estimated by cyclic voltammetry
- [15]. Theoretical calculations and electrochemical properties DFT calculations were employed to gain insight into the structure–property relationships of the quinazoline-based derivatives 1–3. The compounds have phenyl spacers between the donor and acceptor units (Figure 2). Therefore, the dihedral
- results of theoretical calculations which revealed that the HOMO and LUMO orbitals of the carbazole-substituted quinazoline compound 1 overlap. Photophysical and electronic properties Figure 4 shows the theoretical UV spectra and experimental absorption spectra of dilute THF solutions of compounds 1–3









The charge-assisted hydrogen-bonded organic framework (CAHOF) self-assembled from the conjugated acid of tetrakis(4-aminophenyl)methane and 2,6-naphthalenedisulfonate as a new class of recyclable Brønsted acid catalysts
Beilstein J. Org. Chem. 2020, 16, 1124–1134, doi:10.3762/bjoc.16.99

- water and dichloromethane. The methanol and dichloromethane mixture did not need to be considered as F-1 was not soluble in this mixture. For the other two solvent mixtures, the four ammonium groups of the protonated form of TAPM had pKa values in water (according to the calculations discussed above
- providing supercomputer facilities. Funding The X-ray diffraction data were collected with financial support from the Ministry of Science and Higher Education of the Russian Federation using the equipment of the Center for Molecular Composition Studies of INEOS RAS. Quantum chemical calculations were










Synthesis of esters of diaminotruxillic bis-amino acids by Pd-mediated photocycloaddition of analogs of the Kaede protein chromophore
Beilstein J. Org. Chem. 2020, 16, 1111–1123, doi:10.3762/bjoc.16.98

- -squares calculations [70] CCDC-1991019 (2c) and -1991018 (4a) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. General synthesis of the 4-((Z)-arylidene









A cyclopeptide and three oligomycin-class polyketides produced by an underexplored actinomycete of the genus Pseudosporangium
Beilstein J. Org. Chem. 2020, 16, 1100–1110, doi:10.3762/bjoc.16.97

- (10''-NH); HRESITOFMS (m/z): [M − H]− calcd for C36H36N5O7, 650.2615; found, 650.2614. Computational procedure General information Conformational search was performed with MacroModel implemented in the Maestro 11.7 software package [46][47]. All DFT-based calculations were performed with the Gaussian
- . The next optimizations were performed at the B3LYP/6-31G(d) level of theory. Frequency calculations were carried out at the same level of theory to confirm the absence of imaginary frequencies and to obtain thermal corrections to the Gibbs free energies at 1 atm, 298.15 K. The duplicate structures









Synthesis of novel multifunctional carbazole-based molecules and their thermal, electrochemical and optical properties
Beilstein J. Org. Chem. 2020, 16, 1066–1074, doi:10.3762/bjoc.16.93

- ]. ϕFL of compounds 7a and 7b was 5.4% and 97.4%, respectively. An estimated error in quantum yield calculations is ca. 10%. The details of the calculations are given in Supporting Information File 1. Surprisingly, compound 7b, in which the formyl group is at the para position to the carbazole ring, was
- . Supporting Information The Supporting Information features the followings: 1) 1H NMR and 13C NMR spectra; 2) FTIR spectra; 3) mass and HRMS spectra; 4) calculations of relative fluorescence quantum yields. Supporting Information File 294: NMR, FTIR, MS and HRMS spectra of compounds and relative quantum yield
- calculations. Funding This work was supported by the Scientific Research Programme Unit (BAP) of Uludag University [KUAP (F)-2018/14]. Nuray Altinolcek thanks the Scientific and Technological Research Council of Turkey (TÜBİTAK) for a Ph.D. scholarship for domestic priority areas (2211-C). Dr. Ahmet Battal








Aryl-substituted acridanes as hosts for TADF-based OLEDs
Beilstein J. Org. Chem. 2020, 16, 989–1000, doi:10.3762/bjoc.16.88

- . Theoretical calculations The optimized structures of 3–6 were obtained by density functional theory (DFT) calculations at the B3LYP/6-31G(d,p) level of theory (Figure 1). The dihedral angles between the acridanyl and phenyl moieties in compound 3 (37.0 and 36.3°) are comparable with the dihedral angles
- earlier [34]. 1H NMR and 13C NMR spectra were obtained using a Varian Unity Inova (300 MHz (1H) and 75 MHz (13C)). Absorption and photoluminescence (PL) spectra of dilute solutions and of the films were recorded as described previously [35]. Theoretical calculations were carried out using Gaussian 16 [29
- , 29.3, 11.7; MS (APCI+, 20 V) m/z: 426 ([M + H]+); anal. calcd for C29H25F2N: C, 81.86; H, 5.92; F, 8.93; N, 3.29; found: C, 81.91; H, 5.99; N, 3.31%. Theoretically calculated HOMO and LUMO levels distributions and optimized geometries of 3–6 DFT calculations were performed at the B3LYP/6-31G(d,p) level








Synthesis and properties of tetrathiafulvalenes bearing 6-aryl-1,4-dithiafulvenes
Beilstein J. Org. Chem. 2020, 16, 974–981, doi:10.3762/bjoc.16.86

- ; one was the palladium-catalyzed C–H arylation of TTF with bromide 12 (Scheme 2a) and the other was the Vilsmeier–Haack reaction of 1a, followed by triethyl phosphite-mediated cross coupling with 11 (Scheme 2b). Theoretical calculations The DFT calculations of compounds 1a, 3a, and 4 have been carried
- is participating in this redox process is surrounded by extended aromatic rings bearing 1,3-dithiol rings. Conclusion We have synthesized novel multistage TTF derivatives 1–4 bearing 6-aryl-1,4-dithiafulvene moieties by palladium-catalyzed direct C–H arylation. The DFT calculations revealed the








Cation-induced ring-opening and oxidation reaction of photoreluctant spirooxazine–quinolizinium conjugates
Beilstein J. Org. Chem. 2020, 16, 904–916, doi:10.3762/bjoc.16.82

- discussed in the literature whether metal cations are coordinated in a monodentate fashion to the phenolate oxygen atom or rather in a bidentate fashion both to the phenolate oxygen and the imine nitrogen atoms of the open merocyanine form [25][28]. Furthermore, density functional theory (DFT) calculations











Bipyrrole boomerangs via Pd-mediated tandem cyclization–oxygenation. Controlling reaction selectivity and electronic properties
Beilstein J. Org. Chem. 2020, 16, 895–903, doi:10.3762/bjoc.16.81

- using NMR spectroscopy and DFT calculations [33]. In particular, the unprecedented double α-oxygenation of bipyrroles was shown to occur through stepwise acetoxylation, which we found to compete with α–α oligomerization. These new bipyrrole boomerangs exhibited enhanced fluorescence with Φfl values of
- DFT calculations (Figure 1 and Supporting Information File 1). The length of the linker (n) in cNDAnX and cNMInX controls the in- and out-of-plane geometry of the chromophore. The observed changes can be expressed in terms of two parameters: α, the angle between the monopyrrole axis and the N–N vector
- measurements, without any significant loss of the unpolarized emission intensity. This behavior, which precluded a quantitative analysis of the CPL properties, may be attributed to a photoinduced racemization process. The differences in configurational stability of boomerangs are reproduced by DFT calculations





Preparation of 2-phospholene oxides by the isomerization of 3-phospholene oxides
Beilstein J. Org. Chem. 2020, 16, 818–832, doi:10.3762/bjoc.16.75

- double bond migration pathways were elucidated by quantum chemical calculations. Keywords: chlorophosphonium salts; isomerization; 2-phospholene oxides; 3-phospholene oxides; quantum chemistry; Introduction P-Heterocyclic derivatives are valuable targets in synthetic organophosphorus chemistry [1][2][3
- . Moreover, we wish to interpret the different isomerization mechanisms by quantumchemical calculations. Results and Discussion Preparation of 1-substituted-3-methyl-2-phospholene oxides (4) via chlorophospholenium chlorides (2 and 3) The preparation of 2-phospholene oxides 4 was first attempted via the
- for 48 h. Upon this period, nearly complete isomerization of the chloro-3-phospholenium salts 2 to the corresponding chloro-2-phospholenium salts 3 occurred. The mechanism of this isomerization was investigated by quantumchemical calculations in our previous study [65]. However, the formation of the 2







One-pot synthesis of dicyclopenta-fused peropyrene via a fourfold alkyne annulation
Beilstein J. Org. Chem. 2020, 16, 791–797, doi:10.3762/bjoc.16.72

- exhibits a narrow energy gap (1.78 eV) and a lower LUMO energy level than the parent peropyrene without the fusion of the five-membered rings. In addition, the effects of the peri-fused pentagons on the aromaticity and molecular orbitals of 1 were evaluated by theoretical calculations. This work presents
- -membered rings on the peropyrene core, the electronic structures and the frontier orbitals of the peropyrene derivative 6 without pentagons and of compound 1 are compared by DFT calculations at the B3LYP/6-311++G(d,p) level. As shown in Figure 4, the LUMO and HOMO of 6 are both delocalized over the
- -diphenylpyrene is the key step. The single crystal X-ray diffraction analysis revealed a twisted structure of 1 due to the steric hindrance at the bay positions. From the bond length analysis and DFT calculations, CP-PAH 1 consists of the aromatic peropyrene core with two slightly antiaromatic peri-fused five







Towards triptycene functionalization and triptycene-linked porphyrin arrays
Beilstein J. Org. Chem. 2020, 16, 763–777, doi:10.3762/bjoc.16.70

- = 98.157(2)°, V = 1655.53(10) Å3, Z = 4, T = 100(2) K, μ(Cu Kα) = 1.112 mm−1, Dcalc = 1.065 g/cm3, 36162 reflections measured (6.874° ≤ 2θ ≤ 133.814°), 5788 unique (Rint = 0.0696, Rsigma = 0.0442) which were used in all calculations. The final R1 was 0.0808 (I > 2σ(I)) and wR2 was 0.2531 (all data). The
- ) which were used in all calculations. The final R1 was 0.0695 (I > 2σ(I)) and wR2 was 0.1797 (all data). Both phenyl moieties at C15_1 and C15_2 were modelled over two positions in a 56:44% occupancy using the constrain EADP. The porphyrin ring from C17 to C3 in both residues one and two were modelled














Recent advances in Cu-catalyzed C(sp3)–Si and C(sp3)–B bond formation
Beilstein J. Org. Chem. 2020, 16, 691–737, doi:10.3762/bjoc.16.67

- an appropriate electrophile led to C–C bond formation, ultimately delivering chiral tertiary alcohols. Mechanistic studies and DFT calculations showed that an in situ-formed borylcopper(I) species is responsible for the 1,2-addition (Scheme 73) [136]. C,O-Diboration of ketones 464 was explored using

















































































Synthesis of C70-fragment buckybowls bearing alkoxy substituents
Beilstein J. Org. Chem. 2020, 16, 681–690, doi:10.3762/bjoc.16.66

- Laboratory (PAL). The diffraction images were processed by using HKL3000 [29]. Absorption correction was performed with the program PLATON. All the structures were solved by direct methods (SHELXT-2014, 2015 [30] (for 5a, 5b) or XS [31] (for 5c)) and refined by full-matrix least squares calculations on F2









Towards the total synthesis of chondrochloren A: synthesis of the (Z)-enamide fragment
Beilstein J. Org. Chem. 2020, 16, 670–673, doi:10.3762/bjoc.16.64

- of the molecule was elucidated by a combination of NMR, UV and IR spectroscopy and molecular dynamics calculations (MD, MM2) [12]. However, its (Z)-enamide motif and the polyoxygenated middle segment are synthetically challenging. Results and Discussion Synthesis of amide 3 Here we report our





Direct borylation of terrylene and quaterrylene
Beilstein J. Org. Chem. 2020, 16, 621–627, doi:10.3762/bjoc.16.58

- photophysical properties of TB4 are also unchanged by the substituents because the carbon atoms at 2,5,10,13-positions have less coefficients on its HOMO and LUMO, estimated by theoretical calculations. Finally, the same borylation reaction was applied for quaterrylene, resulting in the formation of soluble
- fluorescence at 576 nm with a quantum yield of ΦF = 0.86 at 298 K (Figure 3). Both peaks are slightly red-shifted relative to those of intact terrylene (λabs = 560 nm and λem = 571 nm with ΦF = 0.82 in toluene). We employed density functional theory (DFT) and time-dependent (TD)-DFT calculations, both of them
- calculations showed a good agreement with the observed absorption spectra. Taking the successful result of the terrylene borylation, next we tried to perform the same reaction to quaterrylene (Scheme 2). The quaterrylene was prepared by the oxidative condensation reaction of perylene with TfOH and DDQ [9










Regioselectively α- and β-alkynylated BODIPY dyes via gold(I)-catalyzed direct C–H functionalization and their photophysical properties
Beilstein J. Org. Chem. 2020, 16, 587–595, doi:10.3762/bjoc.16.53

- estimated to be 2.30, 2,14, 2.35, and 2.21 V, respectively, which is consistent with the calculated HOMO–LUMO energy gaps (vide infra, Figure S24, Supporting Information File 1). Theoretical calculations To gain insight into the substitution effect on the electronic properties of the BODIPY derivatives
- , density functional theory (DFT) calculations were performed at the B3LYP/6-31G(d) level of theory. The a2-symmetry of the HOMOs and b2-symmetry of the LUMOs of each, the α- and β-ethynyl-substituted BODIPYs are almost identical to those of the unsubstituted compound 1a (Figure S24, Supporting Information
- , and DFT calculations. Supporting Information File 167: Crystallographic information file of compound 3a. Supporting Information File 168: Crystallographic information file of compound 6b. Acknowledgements We thank Dr. T. Ono at Kyushu University for the help of absolute photoluminescence quantum








Photophysics and photochemistry of NIR absorbers derived from cyanines: key to new technologies based on chemistry 4.0
Beilstein J. Org. Chem. 2020, 16, 415–444, doi:10.3762/bjoc.16.40

- withdrawing moieties. Quantum chemical calculations showed only small contributions of the substituent placed at the meso-position to the electron density in the HOMO/LUMO pattern [81]. Presumably, coupling of lower occupied molecular orbitals with the HOMO on the one hand side and higher unoccupied MOs with























Six-fold C–H borylation of hexa-peri-hexabenzocoronene
Beilstein J. Org. Chem. 2020, 16, 391–397, doi:10.3762/bjoc.16.37

- calculations. The spectra revealed a bathochromic shift of absorption bands compared with unsubstituted HBC under the effect of the σ-donation of boryl groups. Keywords: C–H borylation; hexa-peri-hexabenzocoronene; iridium catalyst; X-ray crystallography; Introduction Polycyclic aromatic hydrocarbons (PAHs
- hexaborylated HBC 1. The structure of thus-obtained 1 was confirmed by X-ray crystallography, and the electronic effects of the boryl groups were investigated through optoelectronic measurements and density functional theory (DFT) calculations. Results and Discussion We have examined the conditions for C–H
- from the singlet excited state were determined (kr = 2.0 × 106 s−1; knr = 7.9 × 107 s−1). The frontier molecular orbitals of 1 calculated by DFT calculations at the B3LYP/6-31G(d) level of theory are shown in Figure 4b. Owing to the highly symmetrical structure of 1, both its HOMO and HOMO−1 as well as




