Search results
Search for "density functional" in Full Text gives 262 result(s) in Beilstein Journal of Organic Chemistry. Showing first 200.
In silico rationalisation of selectivity and reactivity in Pd-catalysed C–H activation reactions
Beilstein J. Org. Chem. 2020, 16, 1465–1475, doi:10.3762/bjoc.16.122
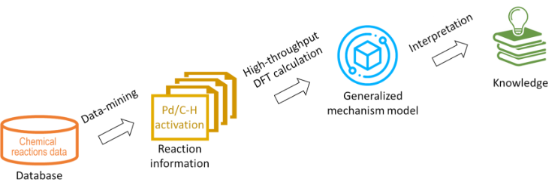
- automated reaction planning, as well as a starting point for microkinetic modelling. Keywords: C–H activation; density functional theory; reaction prediction; Introduction Periodically, our knowledge of chemistry is enriched with new transformations that provide significant breakthroughs by enabling new
- the mechanistic models based on accurate quantum chemical methods, such as the density functional theory (DFT) methods, decreases. Automation of DFT, as well as using results of DFT to develop less expensive predictive models, are the two approaches that may offer the alternatives to the fully data
- transition metal coordination sphere; the energy of a new M–C bond formed and the thermodynamic stability of organometallic product. With new developments in computational chemistry, mechanistic studies using density functional theory (DFT) provide valuable insights into the reactivity of organometallic



Development of fluorinated benzils and bisbenzils as room-temperature phosphorescent molecules
Beilstein J. Org. Chem. 2020, 16, 1154–1162, doi:10.3762/bjoc.16.102

- -deficient fluorinated aromatic ring. To confirm the electron-withdrawing effect of this fluorinated aromatic ring, the electronic charge at the adjacent C≡C bond was calculated by density functional theory (DFT) using the Gaussian 16 (Revision B.01) software package [35]. As typical examples, the molecular
- derivatives previously developed by our group and (B) phosphorescent molecular structures intended for this work. Mulliken charge distributions of fluorinated 1a and nonfluorinated 1c obtained from density functional theory calculations [CAM-B3LYP/6-31+G(d) level]. Absorption and photoluminescence (PL
- molecular orbitals (isosurface value: 0.04 a.u.) involved in vertical electronic transitions in 2a and 3a calculated using density functional theory (DFT) and time-dependent DFT at the CAM-B3LYP/6-31+G(d) level (HOMO: highest occupied molecular orbital, LUMO: lowest unoccupied molecular orbital). Synthetic








Aryl-substituted acridanes as hosts for TADF-based OLEDs
Beilstein J. Org. Chem. 2020, 16, 989–1000, doi:10.3762/bjoc.16.88

- . Theoretical calculations The optimized structures of 3–6 were obtained by density functional theory (DFT) calculations at the B3LYP/6-31G(d,p) level of theory (Figure 1). The dihedral angles between the acridanyl and phenyl moieties in compound 3 (37.0 and 36.3°) are comparable with the dihedral angles








Cation-induced ring-opening and oxidation reaction of photoreluctant spirooxazine–quinolizinium conjugates
Beilstein J. Org. Chem. 2020, 16, 904–916, doi:10.3762/bjoc.16.82

- discussed in the literature whether metal cations are coordinated in a monodentate fashion to the phenolate oxygen atom or rather in a bidentate fashion both to the phenolate oxygen and the imine nitrogen atoms of the open merocyanine form [25][28]. Furthermore, density functional theory (DFT) calculations











Direct borylation of terrylene and quaterrylene
Beilstein J. Org. Chem. 2020, 16, 621–627, doi:10.3762/bjoc.16.58

- fluorescence at 576 nm with a quantum yield of ΦF = 0.86 at 298 K (Figure 3). Both peaks are slightly red-shifted relative to those of intact terrylene (λabs = 560 nm and λem = 571 nm with ΦF = 0.82 in toluene). We employed density functional theory (DFT) and time-dependent (TD)-DFT calculations, both of them










Regioselectively α- and β-alkynylated BODIPY dyes via gold(I)-catalyzed direct C–H functionalization and their photophysical properties
Beilstein J. Org. Chem. 2020, 16, 587–595, doi:10.3762/bjoc.16.53

- , density functional theory (DFT) calculations were performed at the B3LYP/6-31G(d) level of theory. The a2-symmetry of the HOMOs and b2-symmetry of the LUMOs of each, the α- and β-ethynyl-substituted BODIPYs are almost identical to those of the unsubstituted compound 1a (Figure S24, Supporting Information








Six-fold C–H borylation of hexa-peri-hexabenzocoronene
Beilstein J. Org. Chem. 2020, 16, 391–397, doi:10.3762/bjoc.16.37

- -catalyzed six-fold C–H borylation of HBC was successfully achieved by screening solvents. The crystal structure of hexaborylated HBC was confirmed via X-ray crystallography. Optoelectronic properties of the thus-obtained hexaborylated HBC were analyzed with the support of density functional theory
- hexaborylated HBC 1. The structure of thus-obtained 1 was confirmed by X-ray crystallography, and the electronic effects of the boryl groups were investigated through optoelectronic measurements and density functional theory (DFT) calculations. Results and Discussion We have examined the conditions for C–H





p-Pyridinyl oxime carbamates: synthesis, DNA binding, DNA photocleaving activity and theoretical photodegradation studies
Beilstein J. Org. Chem. 2020, 16, 337–350, doi:10.3762/bjoc.16.33

- factor of 1.43 [11]. Theoretical calculations Calculations for the photodegradation of carbamates The structures, properties and the basic photochemistry of compounds 11 and 12 was studied using the density functional theory (DFT) method [86][87][88][89] and the functional B3PW91 along with the 6-31 G(d













The reaction of arylmethyl isocyanides and arylmethylamines with xanthate esters: a facile and unexpected synthesis of carbamothioates
Beilstein J. Org. Chem. 2020, 16, 159–167, doi:10.3762/bjoc.16.18

- reactions, a mechanism is proposed in which the key steps are supported by quantum chemical calculations. Keywords: benzylamines; carbamothioates; density functional theory; intrinsic reaction coordinate analysis; isocyanides; sodium hydride; xanthate esters; Introduction Carbamothioates (thiocarbamates
- , however, carbamothioates 4a–l were instead obtained in 76–88% yield (Scheme 1). Herein, we report on this intriguing finding and show several examples, including a single crystal X-ray structure of one of the products so obtained. A plausible mechanism to explain the reaction using density functional








Synthesis and characterization of bis(4-amino-2-bromo-6-methoxy)azobenzene derivatives
Beilstein J. Org. Chem. 2019, 15, 3000–3008, doi:10.3762/bjoc.15.296

- of intramolecular H-bond formation, we wished to retain at least one methoxy group. To reduce the rate of thermal reversion, only derivatives with substituents in all four ortho-positions were considered. Time-dependent density functional theory (TD-DFT) calculations were used to predict the
- absorption wavelengths of possible derivatives. Based on these considerations, we carried out the synthesis and photochemical characterization of compounds 4 and 5. Results and Discussion Computational chemistry Calculations were performed using density functional theory (DFT) methods (B3LYP/6-31+G**) to






Pigmentosins from Gibellula sp. as antibiofilm agents and a new glycosylated asperfuran from Cordyceps javanica
Beilstein J. Org. Chem. 2019, 15, 2968–2981, doi:10.3762/bjoc.15.293

- ) were not elucidated previously. Therefore, electronic circular dichroism (ECD) measurements combined with time-dependent density functional theory (TDDFT) calculations of ECD data of compound 1 in MeOH at the B3LYP/6-311+G* level of theory were carried out. The CD spectrum of 1 showed strong Cotton







Synthesis and optoelectronic properties of benzoquinone-based donor–acceptor compounds
Beilstein J. Org. Chem. 2019, 15, 2914–2921, doi:10.3762/bjoc.15.285

- carbazoles with respect to the bridging benzene of ca. 44° in both 3 and 4 (Figure 2). Theoretical properties Density functional theory (DFT) calculations were performed in the gas phase to assess the electronic structures of 2–5 (see Supporting Information File 1 for details). The S1 and T1 excited states
- were calculated from the optimized ground-state structure using the Tamm Dancoff approximation (TDA) to time-dependent density functional theory (TD-DFT) [30][31]. In all derivatives, the LUMO is mainly localized on the strongly electron-accepting BQ moiety (Figure 3). The HOMO is mainly located on the










Carbazole-functionalized hyper-cross-linked polymers for CO2 uptake based on Friedel–Crafts polymerization on 9-phenylcarbazole
Beilstein J. Org. Chem. 2019, 15, 2856–2863, doi:10.3762/bjoc.15.279

- from the adsorption branches using nonlocal density functional theory (NLDFT) methods. P2–P5 exhibited narrow pore size distribution in the micropore scopes (<2 nm), while P1 showed a wide pore size distribution. The surface areas of P1–P5 ranged from 350 to 769 m2 g−1 (Table 2). The lowest specific









A combinatorial approach to improving the performance of azoarene photoswitches
Beilstein J. Org. Chem. 2019, 15, 2753–2764, doi:10.3762/bjoc.15.266

- performance: o-F [13][14], o-Cl [8][29][30], o-OMe [10][29][31], or o-Pyr [32]. For the sake of comparison, both mono- and di-ortho-substitutions were considered. Theoretical half-lives (t1/2) were calculated within the density functional theory (DFT) framework, according to the protocol reported in our prior
- ground state geometries of both Z- and E-isomers were computed under the time-dependent density functional theory (TDDFT) approach [41][42]. The 20 lowest-lying singlet excited states were calculated in all conformers at the TD-PBE0/6-31G** level of theory in the gas phase. Solvent effects in









Acid-catalyzed rearrangements in arenes: interconversions in the quaterphenyl series
Beilstein J. Org. Chem. 2019, 15, 2655–2663, doi:10.3762/bjoc.15.258

- . This supports thermodynamic control based on carbocation energies. Keywords: arenium ion; carbocation; density functional theory; microwave reaction; rearrangement; superacid; Introduction Carbocations are enigmatic reactive intermediates of enduring importance in chemistry. No other reactive species







Plasma membrane imaging with a fluorescent benzothiadiazole derivative
Beilstein J. Org. Chem. 2019, 15, 2644–2654, doi:10.3762/bjoc.15.257

- density functional theory (TD-DFT). In practice, when applying DFT calculations, there is no “universal” exchange correlation functional (XCF), thus the performance of different XCFs in simulating the absorption spectra of BTD-4APTEG had to be assessed. We aimed at describing the maxima absorption peak
- were carried out with the long-range corrected density functional CAM-B3LYP with 6-31G(d) Pople’s split basis set. Harmonic frequency calculations were performed to verify that a genuine energetic minimum was achieved. Solvent effects on the BTD-4APTEG geometries were assessed using the polarizable









Experimental and computational electrochemistry of quinazolinespirohexadienone molecular switches – differential electrochromic vs photochromic behavior
Beilstein J. Org. Chem. 2019, 15, 2473–2485, doi:10.3762/bjoc.15.240

- cyclic voltammetry and 1H NMR analyses, in concert with computational modeling. These results are compared to those for the symmetric parent PSHD, which due to symmetry possesses only a single possible regioisomer upon either electrochemical or photochemical ring-opening. Density functional theory
- calculations of bond lengths, bond orders, and molecular orbitals allow the rationalization of this differential photochromic vs electrochromic behavior of the QSHDs. Keywords: cyclic voltammetry; density functional theory; heterocycles; molecular switches; photochromic photooxidants; spirocycles
- comparing orbitals of SW* to those of SW and SW•−. Using S1 for SW* would require unbalanced ground and excited-state calculations, e.g., time-independent and time-dependent density functional theories, or single-reference and multi-reference methods. However, using T0 for SW* is a straightforward ground











Reversible switching of arylazopyrazole within a metal–organic cage
Beilstein J. Org. Chem. 2019, 15, 2398–2407, doi:10.3762/bjoc.15.232

- -ray diffraction. To obtain hints about the packing of Z-1 within 2, we therefore performed density functional theory (DFT) calculations. Compared with the complex before photoisomerization, the cage in (Z-1)2 is severely distorted (Figure 5; see also Supporting Information File 3), assuming a bowl



![[Graphic 1]](/bjoc/content/inline/1860-5397-15-232-i3.svg?max-width=637&scale=1.18182) 2 (500 MHz, D2O, 298 K).
2 (500 MHz, D2O, 298 K).







Excited state dynamics for visible-light sensitization of a photochromic benzil-subsituted phenoxyl-imidazolyl radical complex
Beilstein J. Org. Chem. 2019, 15, 2369–2379, doi:10.3762/bjoc.15.229

- chromatography (HPLC), and each isomer was characterized by steady-state absorption spectra and time-dependent density functional theory (TDDFT) calculations as shown below. Figure 1 shows the steady-state absorption spectra of the two isomers of Benzil-PIC and PIC in benzene at 298 K. While the absorption of








Current understanding and biotechnological application of the bacterial diterpene synthase CotB2
Beilstein J. Org. Chem. 2019, 15, 2355–2368, doi:10.3762/bjoc.15.228

- rearrangement. Hong and Tantillo performed the first theoretical study of the reaction mechanism in CotB2 via gas phase density functional theory (DFT) calculations [33]. Simultaneously, Sato et al. also studied the CotB2 reaction mechanism in the gas phase using DFT, combined with experimental deuterium
- ], density functional theory calculations [33] as well as QM/MM simulations [37]. Variants of CotB2 open the route to a novel product portfolio with altered cyclic carbon skeletons, which can be converted into bioactive compounds by chemo-enzymatic methodologies. Modification descriptions are composed











Aggregation-induced emission effect on turn-off fluorescent switching of a photochromic diarylethene
Beilstein J. Org. Chem. 2019, 15, 2204–2212, doi:10.3762/bjoc.15.217

- decreased gradually upon UV light irradiation accompanied with the formation of 1c, because of excitation energy transfer from the ESIPT moiety to the closed-ring isomer (Figure 5) [4]. The wavelengths of absorption (Table 3) and fluorescence (Table 4) were obtained computationally by using density
- functional theory (DFT) and time-dependent DFT (TDDFT). The excitation wavelengths as well as the emission wavelength qualitatively agrees with the experimental results. Since compound 1 consists of a diarylethene moiety and an imidazo[1,2-a]pyridine moiety, the characteristic of 1 has the combination of








Azologization and repurposing of a hetero-stilbene-based kinase inhibitor: towards the design of photoswitchable sirtuin inhibitors
Beilstein J. Org. Chem. 2019, 15, 2170–2183, doi:10.3762/bjoc.15.214

- ortho methyl groups in 2f, intramolecular photocyclization could be prevented. To verify the hypothetical structures derived from irradiation of 2b, we carried out quantum chemical calculations of the double bond isomers (E)-2b and (Z)-2b as well as the oxidized compounds 8a and 8b. We used density
- functional theory (DFT) to optimize the ground state equilibrium structures of (E)-2b, (Z)-2b, 8a and 8b, and used time-dependent DFT (TDDFT) and high-level correlated methods to obtain UV–vis absorption energies and oscillator strengths. To obtain the simulated absorption spectrum and λmax values










1,2,3,4-Tetrahydro-1,4,5,8-tetraazaanthracene revisited: properties and structural evidence of aromaticity loss
Beilstein J. Org. Chem. 2019, 15, 2059–2068, doi:10.3762/bjoc.15.203

- level optimized and experimental geometry of THTAA and its derivatives show considerable loss of aromaticity within the quinoxaline moiety. Keywords: aromaticity; density functional calculations; heterocycles; hydrogen bonds; X-ray structures; Introduction Quinoxaline derivatives 1 – also called















Bipolenins K–N: New sesquiterpenoids from the fungal plant pathogen Bipolaris sorokiniana
Beilstein J. Org. Chem. 2019, 15, 2020–2028, doi:10.3762/bjoc.15.198

- -13 were α-oriented. The absolute configuration of 1 was determined to be 1R,3R,6S,7R,13S by comparison of the experimental electronic circular dichroism (ECD) spectrum with time-dependent density functional theory (TDDFT)-calculated ECD spectra of the two possible enantiomers of 1 (Figure 4






A review of the total syntheses of triptolide
Beilstein J. Org. Chem. 2019, 15, 1984–1995, doi:10.3762/bjoc.15.194

- -endo-trig cyclization of 2-alkenyl-1,3-dithiolanes to access trans-decalins (Figure 2, route K) [78]. Density functional theory calculation (DFT) studies indicated that the 2-alkenyl-1,3-dithiolane moiety acts as a latent initiator, which triggers the cationic 6-endo-trig cyclization in the presence of










