Search results
Search for "docking" in Full Text gives 75 result(s) in Beilstein Journal of Organic Chemistry.
Synthesis and biological evaluation of truncated derivatives of abyssomicin C as antibacterial agents
Beilstein J. Org. Chem. 2019, 15, 1468–1474, doi:10.3762/bjoc.15.147

- docking of known and proposed ligands. The crystal structure contains a tryptophan molecule in the active site. Restrained molecular dynamics [20][21] was employed to position the active site cysteine (Cys-263) in a position that would allow covalent binding of the ligands in the active site. The
- resulting protein conformation was allowed to relax to ensure that the found conformation was an energy minimum. This protein structure was then used for docking of ligands using Glide [22][23][24]. Glide XP docking positioned both AbC and the analog atrop-O-benzyl-desmethyl-abyssomicin C with low energy
- conformations and with expected interaction points in the binding site, providing suitable docking scores (see Supporting Information File 1). The key interactions include a hydrogen bond to the backbone of Arg-45 and lipophilic interactions in the deep pocket defined by Phe-241 and Leu-34 (Figure 2). Our







Towards the preparation of synthetic outer membrane vesicle models with micromolar affinity to wheat germ agglutinin using a dialkyl thioglycoside
Beilstein J. Org. Chem. 2019, 15, 937–946, doi:10.3762/bjoc.15.90

- 8 showed the highest inhibitory effect of this study with an IC50 of 11 µM corresponding to a 3000-fold improvement compared to the monomer control. This result suggests a higher participation of sugar units in the lectin binding for this compound. Docking calculation for model glycolipids To
- appreciate the possible binding of 4, 5, 7 and 8 onto WGA, a docking simulation was performed using the crystal structure of WGA obtained in the presence of GlcNAc (PDB 2UVO) and of other GlcNAc derivatives (PDB 2X52 and 4AML). The docking experiments were performed using analogous molecules bearing acetate
- and butanoate alkyl chains (namely 4C2, 4C4, 5C2, 5C4, 7C2, 7C4, 8C2 and 8C4 according to the alkyl length, see Figures S5 and S6 in Supporting Information File 1) instead of the long alkyl chains, for calculation simplicity. The docking protocol was validated using GlcNAc and Glc as ligands. In such








![[Graphic 2]](/bjoc/content/inline/1860-5397-15-90-i4.svg?max-width=637&scale=0.472728) ) and (○) ...
) and (○) ...

Stereochemical investigations on the biosynthesis of achiral (Z)-γ-bisabolene in Cryptosporangium arvum
Beilstein J. Org. Chem. 2019, 15, 789–794, doi:10.3762/bjoc.15.75

- resulting in (S)-A after ring closure (Scheme 1, path A) or via (S)-NPP, which would suggest involvement of (R)-A (Scheme 1, path B). This stereochemical link between NPP and A was also observed in a theoretical docking study with epi-isozizaene synthase suggesting (S)-NPP and (R)-A to be included in its






Back to the future: Why we need enzymology to build a synthetic metabolism of the future
Beilstein J. Org. Chem. 2019, 15, 551–557, doi:10.3762/bjoc.15.49

- screened for new catalytic reactions in metabolic retrosynthesis. Further improvements in homology modeling and virtual docking are expected to increase accuracy and throughput, which will help to map and predict the substrate and reactions catalyzed by an enzyme superfamily and its individual members in



Design of indole- and MCR-based macrocycles as p53-MDM2 antagonists
Beilstein J. Org. Chem. 2019, 15, 513–520, doi:10.3762/bjoc.15.45

- -pocket as shown by our docking studies (Figure 1A,B, Figure S4 in Supporting Information File 1). Thus, extending our previous work [13], the Leu26 subpocket was probed by utilizing the different ring sizes and the different heteroatoms (oxygen or sulfur) of our macrocyclic library. In addition, the






Protein–protein interactions in bacteria: a promising and challenging avenue towards the discovery of new antibiotics
Beilstein J. Org. Chem. 2018, 14, 2881–2896, doi:10.3762/bjoc.14.267

- was screened by X-ray crystallography leading to the identification of four fragment hits. In an attempt to improve their binding affinities, another library was searched for compounds displaying similarity to these initial hits. After a docking-based screening followed by a fluorescence polarization
- database [82] was searched for structurally similar compounds leading to the identification of the meta-substituted tetrazole 33 (Figure 7), which was found to have a similar dissociation constant. Moreover, in order to predict the orientation of the fragments in the binding site, molecular docking of 33










Targeting the Pseudomonas quinolone signal quorum sensing system for the discovery of novel anti-infective pathoblockers
Beilstein J. Org. Chem. 2018, 14, 2627–2645, doi:10.3762/bjoc.14.241

- docking [51]. These inhibitors appeared to bind in the substrate channel in a slightly remote position from the active site cysteine and, hence, termed channel blockers [51]. Optimised hits exhibited a potency in the single-digit micromolar range (12, Figure 6). However, it has been found that similar
- tailor-made SPR experiment including truncated and elongated derivatives as well as nitrophenylmethanol-based active-site blockers of different size as competitors. These experiments combined with molecular docking (Figure 7) led to the postulation of a plausible binding pose characterising the
- , their potency in attenuating biofilm formation was more pronounced than ciprofloxacin and linezolid itself. A docking study suggested PqsD to be the target of these compounds like 23 (Figure 10), although this remains speculative. PqsE inhibitors The pathway-specific thioesterase PqsE is not only
























Microwave-assisted synthesis of biologically relevant steroidal 17-exo-pyrazol-5'-ones from a norpregnene precursor by a side-chain elongation/heterocyclization sequence
Beilstein J. Org. Chem. 2018, 14, 2589–2596, doi:10.3762/bjoc.14.236

- hormones, and therefore, in the development of prostate cancer [1]. According to extensive structure–activity relationship and docking studies, a potent steroidal inhibitor should possess certain structural characteristics for efficient P45017α inhibition [1][2][3], such as (i) a five or six-membered non




Synthesis of 3-aminocoumarin-N-benzylpyridinium conjugates with nanomolar inhibitory activity against acetylcholinesterase
Beilstein J. Org. Chem. 2018, 14, 2545–2552, doi:10.3762/bjoc.14.231

- potent activities with inhibitory concentration (IC50) values in the nanomolar concentration range. Among them, the 2,3-difluorobenzylpyridinium-containing compound was the most potent inhibitor with an IC50 value of 1.53 ± 0.01 nM. Docking studies revealed that the synthesized compounds inhibit the
- well as with benzylpyridinium moieties [12][13][14] have been reported as dual-binding site AChE inhibitors. Recently we have reported the AChE inhibitory activity of the coumarin derivative, scopoletin conjugated with a pyridinium side chain (Figure 1) [15] and a docking study revealed that the
- that may be useful for binding with this enzyme. Herein, we report our progress on the synthesis, biological evaluation, and molecular docking of 3-aminocoumarin linked with the benzylpyridinium moiety through an amide bond. Results and Discussion Chemistry The target 3-aminocoumarin-N-benzylpyridinium





The enzymes of microbial nicotine metabolism
Beilstein J. Org. Chem. 2018, 14, 2295–2307, doi:10.3762/bjoc.14.204

- the crystal structure of the enzyme confirmed these conclusions (Figure 2) [27]. Docking of (R)-6-hydroxynicotine into the structure yielded a model for substrate binding. Vanillyl oxidase catalyzes the oxidation of 4-hydroxybenzyl alcohols, the oxidative deamination of 4-hydroxybenzylamines, and the
















Diazirine-functionalized mannosides for photoaffinity labeling: trouble with FimH
Beilstein J. Org. Chem. 2018, 14, 1890–1900, doi:10.3762/bjoc.14.163

- accuracy. We are concluding from this study that photolabeling of FimH with sugar diazirines has only very limited success and cannot be regarded a facile approach for covalent modification of FimH. Keywords: diazirines; docking; FimH; lectin ligands; mannosides; mass spectrometry; photoaffinity labelling
- mannosides 3–5 as photolabile ligands of FimH and evaluated their potential affinity by computer-aided docking. Docking studies Mannosides 3–5 (Figure 3) were docked into the FimH carbohydrate binding site using FlexX as implemented in Sybyl 6.9 [21][22][23]. Ligand structures were minimized using the Tripos
- carbohydrate binding site. They are flexible and can be more distant to one another (“open gate”) or closer together (“closed gate”) [20]. Both conformations were considered for the docking studies. For each docked conformation, a scoring value is obtained that correlates with the affinity of the ligand to the








The phenyl vinyl ether–methanol complex: a model system for quantum chemistry benchmarking
Beilstein J. Org. Chem. 2018, 14, 1642–1654, doi:10.3762/bjoc.14.140

- -docking motifs via the phenyl and vinyl moieties, with an additional less populated OH∙∙∙P(phenyl)-bound isomer detected only by microwave spectroscopy. The correct prediction of the energetic order of the isomers using quantum-chemical calculations turns out to be challenging and succeeds with a
- sophisticated local coupled cluster method. The latter also yields a quantification as well as a visualization of London dispersion, which prove to be valuable tools for understanding the role of dispersion on the docking preferences. Beyond the structural analysis of the electronic ground state (S0), the
- and the respective role of different intermolecular forces such as electrostatic, dispersion and induction forces is needed. Thus, experimental examination as well as the precise prediction of a preferred molecular docking site for different molecules is of crucial importance. Despite the remarkable







Novel unit B cryptophycin analogues as payloads for targeted therapy
Beilstein J. Org. Chem. 2018, 14, 1281–1286, doi:10.3762/bjoc.14.109

- glycol spacer with a terminal azide results in a complete loss of activity. Docking studies of the novel cryptophycin analogues to β-tubulin provided a rationale for the observed cytotoxicities. Keywords: cryptophycin; cytotoxic agents; novel payloads; tubulin inhibitors; tumour targeting; Introduction
- appropriate stability and activity to the future conjugate. Results and Discussion Design and synthesis Previous docking studies have postulated that the methyl group of unit B is not involved in the cryptophycin–tubulin interaction [52]. Moreover, its absence did not produce a dramatic loss of activity [24
- activity of analogue 22 could be due to its poor internalization or the modification could alter the interaction with tubulin. In order to get an extensive knowledge of the novel analogues, we embarked in docking and modelling studies, herein reported, and internalization studies are ongoing in our







An overview of recent advances in duplex DNA recognition by small molecules
Beilstein J. Org. Chem. 2018, 14, 1051–1086, doi:10.3762/bjoc.14.93
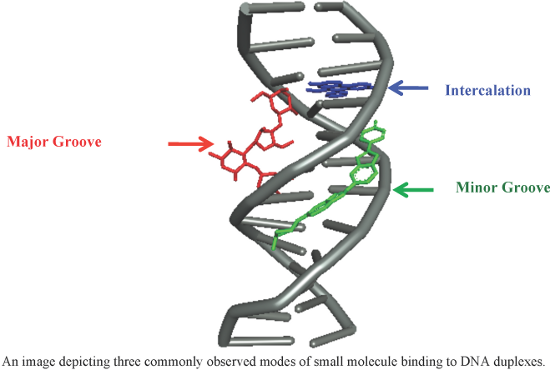
- absorption spectroscopy, viscosity measurement, cyclic voltammetry and molecular modeling (Figure 9) [93]. Docking results confirmed that these complexes have the ability to interact with the minor groove of the ct-DNA. In addition, the authors confirmed that in presence of ascorbic acid, these complexes
- activities were evaluated [102]. A molecular docking study has revealed that GRA interacts with ct-DNAs via hydrogen bonding interactions between the oxygen atoms of GRA and adenine bases of DNA and van der Waals interactions. Moreover, GRA significantly reduces the polymerization activity of DNA polymerase






















Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids – tutorial
Beilstein J. Org. Chem. 2018, 14, 84–105, doi:10.3762/bjoc.14.5
















15N-Labelling and structure determination of adamantylated azolo-azines in solution
Beilstein J. Org. Chem. 2017, 13, 2535–2548, doi:10.3762/bjoc.13.250

- studies and computer-assisted drug design, e.g., molecular docking techniques. Thus, the development of effective methods for the unambiguous determination of N-alkylation site(s) in the azolo-azine series is important. The data that are required to solve this problem could be provided by 15N NMR











Aqueous semisynthesis of C-glycoside glycamines from agarose
Beilstein J. Org. Chem. 2017, 13, 1222–1229, doi:10.3762/bjoc.13.121

- aminomonosaccharides 9 and 13 are interesting moieties, regarding their resemblance with the bioactive (+)-muscarine 14 (Figure 2). Indeed, ongoing docking and molecular dynamics experiments revealed the amino-AnGal moiety as a promising platform to launch the design of new mAChR modulators [11]. The differences in





Strategies in megasynthase engineering – fatty acid synthases (FAS) as model proteins
Beilstein J. Org. Chem. 2017, 13, 1204–1211, doi:10.3762/bjoc.13.119

- in ACP docking and acyl-moiety binding, and allowed catching an initial glimpse of the dynamic process of ACP substrate delivery. Also the interaction of ACP VinL and the acyltransferase VinK, involved in loading a PKS megasynthase, was recently resolved in structure [38]. It is reasonable to assume
- shown for clarity. Each ACP acts in cis for substrate elongation (see Figure 2b) and in trans for substrate translocation. Strategies of megasynthase engineering. a) Mix-and-match approach: A hypothetical chimeric PKS is assembled module by module from a pool of available PKS. In such approach, docking





Synthesis of spiro[isoindole-1,5’-isoxazolidin]-3(2H)-ones as potential inhibitors of the MDM2-p53 interaction
Beilstein J. Org. Chem. 2016, 12, 2793–2807, doi:10.3762/bjoc.12.278

- be linked to the inhibition of the protein–protein p53-MDM2 interaction. Docking measurements support the biological data. Keywords: antitumor agents; DFT studies; 1,3-dipolar cycloaddition; docking studies; spiro-compounds; Introduction The p53 tumor suppressor protein is a transcriptional factor
- -ethynylbenzamides 1 [35]. The rationale of our choice is based on molecular docking data. Using the published structure of the MDM2–p53 binding site, we have employed computational methods and focused library synthesis based on the isoindolinone template, to develop compounds with inhibitory activity. These studies
- 11c). These set of experiments demonstrate that the exposure of SH-SY5Y cancer cell lines to 10 µM 6e for 72 h was able to activate the apoptotic pathway. Docking studies To support the suggested interaction of synthesized compounds with MDM2, docking studies were applied, starting from the X-ray

















Chemical probes for competitive profiling of the quorum sensing signal synthase PqsD of Pseudomonas aeruginosa
Beilstein J. Org. Chem. 2016, 12, 2784–2792, doi:10.3762/bjoc.12.277

- decrease in the production of the virulence factors pyocyanine and pyoverdine [32]. So far only laborious enzyme-based assays, docking studies or modelling resulted in new scaffolds. We were thus interested, if our probes could be applied as a simple tool to discover novel scaffolds or chemical PqsD







Computational methods in drug discovery
Beilstein J. Org. Chem. 2016, 12, 2694–2718, doi:10.3762/bjoc.12.267

- methods are discussed. Advances in virtual high-throughput screening, protein structure prediction methods, protein–ligand docking, pharmacophore modeling and QSAR techniques are reviewed. Keywords: ADME; computer-aided drug design; docking; free energy; high-throughput screening; LBDD; lead optimization
- it possible to use structure-based tools such as virtual high-throughput screening and direct docking methods on targets and possible drug molecules. The affinity of molecules to targets can be evaluated by computing various estimates of binding free energies. Further filtering and optimization of
- structure prediction tools that are routinely used in structure-based drug discovery, widely used docking algorithms, scoring functions, virtual high-throughput screening, lead optimization and methods of assessment of ADME properties of drugs. Review Structure-based drug discovery (SBDD) If the three










Beta-hydroxyphosphonate ribonucleoside analogues derived from 4-substituted-1,2,3-triazoles as IMP/GMP mimics: synthesis and biological evaluation
Beilstein J. Org. Chem. 2016, 12, 1476–1486, doi:10.3762/bjoc.12.144

- -containing derivatives are biologically interesting (Figure 1) [7]. In addition, molecular docking studies have been performed and highlighted the importance of three binding areas within the active site of the protein: a hydrophobic clamp (Phe157, His209 and Tyr210) interacting with the nucleobase, a
- which they block the enzyme activity. For this purpose, a SAR study was carried out by molecular docking. Attempts to make a direct correlation between the inhibitory activity and the gold-computed docking score (Table 2) were unsuccessful. Indeed, all compounds exhibited a similar score with a value
- substituent varies mainly in size, the docking score was normalized by dividing it by the number of heavy atoms (NHA, non-hydrogen atoms). By this mean and in respect to IMP (used as a control as it is the natural substrate of cN-II) one group of derivatives is predicted as good binder (1n, 1o, 1p and 1q











Discovery of an inhibitor of the production of the Pseudomonas aeruginosa virulence factor pyocyanin in wild-type cells
Beilstein J. Org. Chem. 2016, 12, 1428–1433, doi:10.3762/bjoc.12.137

- order to further explore the possibility that compound 4 may act as a LasR antagonist, it was subjected to molecular docking studies against the P. aeruginosa LasR ligand–binding domain (LBD) [31]. Specifically, both OdDHL and 4 were docked into the OdDHL binding pocket of two LasR LBD structures, one
- with a bridging water molecule, which is known to be involved in a hydrogen bonding between OdDHL and Arg61 (Figure 3a) [32], and one without. In addition, both rigid and flexible conformations of LasR LBD were used in the docking runs. Tyr47 and Arg61 exhibit considerable variation in their side-chain
- conformations in various crystal structures of LasR LBD complexes and hence, they were made flexible in the flexible receptor docking runs. The best score for OdDHL (−9.1 kcal/mol) was obtained from the rigid receptor docking run with water. The docked and crystallographic conformations of OdDHL agree closely





Cyclisation mechanisms in the biosynthesis of ribosomally synthesised and post-translationally modified peptides
Beilstein J. Org. Chem. 2016, 12, 1250–1268, doi:10.3762/bjoc.12.120

- ” was a zinc-containing cyclase, and the “D-protein” possesses ATPase activity. The requirement for ATP turnover during cyclisation led to the hypothesis that the D-protein was a docking protein that regulates heterocyclase activity [33], while the presence of zinc in the C-protein pointed towards a












Is conformation a fundamental descriptor in QSAR? A case for halogenated anesthetics
Beilstein J. Org. Chem. 2016, 12, 760–768, doi:10.3762/bjoc.12.76

- ], but these are mostly complementary and are aimed at corroborating and/or rationalizing the results provided by the regression models, since the docking methodology itself provides intermolecular energies and docking scores that correlate with bioactivity. On the other hand, despite not encoding




