Search results
Search for "theoretical calculations" in Full Text gives 120 result(s) in Beilstein Journal of Organic Chemistry.
Conformational preferences of fluorine-containing agrochemicals and their implications for lipophilicity prediction
Beilstein J. Org. Chem. 2020, 16, 2469–2476, doi:10.3762/bjoc.16.200

- conformers (II, VI, and VII), and with a rotatable C–C(F) bond that generates different conformers (I). The μ values for all herbicides were computed through theoretical calculations (see computational details section) and are presented in Table 2 along with their respective experimental log P data








Styryl-based new organic chromophores bearing free amino and azomethine groups: synthesis, photophysical, NLO, and thermal properties
Beilstein J. Org. Chem. 2020, 16, 2282–2296, doi:10.3762/bjoc.16.189

- little effect on the shift for dye 3 (6 nm). In case of the Schiff bases 8–11, similar bathochromic shifts were observed. In addition, experimentally, there was no solvatochromic behavior observed for dyes 7 and 12 as was predicted by theoretical calculations. Based on theoretical calculations, the main












Naphthalene diimide–amino acid conjugates as novel fluorimetric and CD probes for differentiation between ds-DNA and ds-RNA
Beilstein J. Org. Chem. 2020, 16, 2032–2045, doi:10.3762/bjoc.16.170

- with significant Stokes shifts (+60 nm) and quantum yields of 10–32% (Table 1). Theoretical calculations To get insight into the electronic and optical properties of the 2-amino-6-chloro-substituted NDIs, we carried out computational investigations by using the Gaussian 09 program suite [32]. In










One-pot synthesis of oxazolidinones and five-membered cyclic carbonates from epoxides and chlorosulfonyl isocyanate: theoretical evidence for an asynchronous concerted pathway
Beilstein J. Org. Chem. 2020, 16, 1805–1819, doi:10.3762/bjoc.16.148
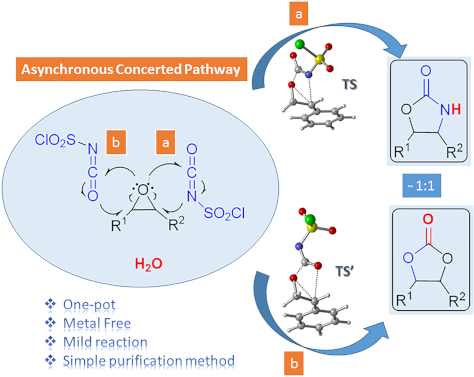
- reaction of CSI with epoxides in different solvents under mild conditions and compared the reaction mechanism with previously proposed mechanisms using theoretical calculations. Keshava Murthy and Dhar [41] postulated a mechanism involving a zwitterionic intermediate. C–O bond cleavage in this unstable and















Recent synthesis of thietanes
Beilstein J. Org. Chem. 2020, 16, 1357–1410, doi:10.3762/bjoc.16.116

- diradicals as intermediates. To explain the regioselectivity, theoretical calculations were performed with thiochalcone and acrylonitrile as model substrates. For the frontier molecular orbital treatment, the largest coefficients in both HOMO and LUMO of thiochalcone existed on the sulfur atom, while the












































































































































Highly selective Diels–Alder and Heck arylation reactions in a divergent synthesis of isoindolo- and pyrrolo-fused polycyclic indoles from 2-formylpyrrole
Beilstein J. Org. Chem. 2020, 16, 1320–1334, doi:10.3762/bjoc.16.113

- ]. Theoretical calculations The Diels–Alder reaction of vinylpyrroles 8b, 8c and 8g with maleimides 7b and 7c resulted in a highly diastereoselective cycloaddition leading to the mixture of endo/exo cycloadducts 9/10, where the endo product 9 was the major one (Scheme 3 and Table 2). Considering the synthetic
- pyrrolizine-containing polycycles, including isoindolo[2,1-a]indole-based tetrahydro and fully aromatic pentacycles. These findings reveal the synthetic value of 2-formylpyrrole (13a) for the diastereo- and regioselective construction of isoindolo- and pyrrolo-fused polycyclic indoles. Theoretical
- calculations suggest that the greater stability of the endo TSs in the Diels–Alder cycloadditions is associated with NCIs between the N-substituents of both cycloaddends. These interactions are also involved in the case of pyrrolo[2,1-a]isoindole 18a with dienophile 7c. Fused aza-hetero polycyclic frames and













Development of fluorinated benzils and bisbenzils as room-temperature phosphorescent molecules
Beilstein J. Org. Chem. 2020, 16, 1154–1162, doi:10.3762/bjoc.16.102

- corresponding bis-oxidized bisbenzil derivatives. Based on theoretical calculations, the selective formation of the fluorinated analogues stemmed from the slight modulation of the charge distribution at the alkyne moiety of the reactant induced by the electron-withdrawing fluorine atoms. Evaluation of the








Synthesis and properties of quinazoline-based versatile exciplex-forming compounds
Beilstein J. Org. Chem. 2020, 16, 1142–1153, doi:10.3762/bjoc.16.101

- compounds, based on theoretical calculations and experimental measurements, as well as the electrochemical and thermal properties, are discussed. The synthesized compounds form glasses with glass-transition temperatures ranging from 116 °C to 123 °C. The ionization potentials estimated by cyclic voltammetry
- [15]. Theoretical calculations and electrochemical properties DFT calculations were employed to gain insight into the structure–property relationships of the quinazoline-based derivatives 1–3. The compounds have phenyl spacers between the donor and acceptor units (Figure 2). Therefore, the dihedral
- results of theoretical calculations which revealed that the HOMO and LUMO orbitals of the carbazole-substituted quinazoline compound 1 overlap. Photophysical and electronic properties Figure 4 shows the theoretical UV spectra and experimental absorption spectra of dilute THF solutions of compounds 1–3









Aryl-substituted acridanes as hosts for TADF-based OLEDs
Beilstein J. Org. Chem. 2020, 16, 989–1000, doi:10.3762/bjoc.16.88

- . Theoretical calculations The optimized structures of 3–6 were obtained by density functional theory (DFT) calculations at the B3LYP/6-31G(d,p) level of theory (Figure 1). The dihedral angles between the acridanyl and phenyl moieties in compound 3 (37.0 and 36.3°) are comparable with the dihedral angles
- earlier [34]. 1H NMR and 13C NMR spectra were obtained using a Varian Unity Inova (300 MHz (1H) and 75 MHz (13C)). Absorption and photoluminescence (PL) spectra of dilute solutions and of the films were recorded as described previously [35]. Theoretical calculations were carried out using Gaussian 16 [29








Synthesis and properties of tetrathiafulvalenes bearing 6-aryl-1,4-dithiafulvenes
Beilstein J. Org. Chem. 2020, 16, 974–981, doi:10.3762/bjoc.16.86

- ; one was the palladium-catalyzed C–H arylation of TTF with bromide 12 (Scheme 2a) and the other was the Vilsmeier–Haack reaction of 1a, followed by triethyl phosphite-mediated cross coupling with 11 (Scheme 2b). Theoretical calculations The DFT calculations of compounds 1a, 3a, and 4 have been carried








One-pot synthesis of dicyclopenta-fused peropyrene via a fourfold alkyne annulation
Beilstein J. Org. Chem. 2020, 16, 791–797, doi:10.3762/bjoc.16.72

- exhibits a narrow energy gap (1.78 eV) and a lower LUMO energy level than the parent peropyrene without the fusion of the five-membered rings. In addition, the effects of the peri-fused pentagons on the aromaticity and molecular orbitals of 1 were evaluated by theoretical calculations. This work presents
- -diphenylethyne, Pd2(dba)3, P(o-tol)3, KOAc, LiCl, DMF, 130 °C, microwave, 6 h, 5%. Supporting Information Supporting Information File 276: Experimental details, synthetic procedures, single crystal X-ray data for 1, detailed theoretical calculations, and analytical data for the compounds. Acknowledgements We







Direct borylation of terrylene and quaterrylene
Beilstein J. Org. Chem. 2020, 16, 621–627, doi:10.3762/bjoc.16.58

- photophysical properties of TB4 are also unchanged by the substituents because the carbon atoms at 2,5,10,13-positions have less coefficients on its HOMO and LUMO, estimated by theoretical calculations. Finally, the same borylation reaction was applied for quaterrylene, resulting in the formation of soluble










Regioselectively α- and β-alkynylated BODIPY dyes via gold(I)-catalyzed direct C–H functionalization and their photophysical properties
Beilstein J. Org. Chem. 2020, 16, 587–595, doi:10.3762/bjoc.16.53

- estimated to be 2.30, 2,14, 2.35, and 2.21 V, respectively, which is consistent with the calculated HOMO–LUMO energy gaps (vide infra, Figure S24, Supporting Information File 1). Theoretical calculations To gain insight into the substitution effect on the electronic properties of the BODIPY derivatives








p-Pyridinyl oxime carbamates: synthesis, DNA binding, DNA photocleaving activity and theoretical photodegradation studies
Beilstein J. Org. Chem. 2020, 16, 337–350, doi:10.3762/bjoc.16.33

- ∙1010 s−1, which is a very large value, indicating a fast N−O bond dissociation for compound 12. Equations 1–3 are described in the theoretical calculations section. As soon as the two radicals (amidinyl and p-chlorocarbamoyloxyl) are formed, the second radical starts to decarboxylate according to the
- chemical reaction below (Scheme 3). The activation free energy for the decarboxylation reaction is only 1.09 kcal/mol and by using Equation 4 (see theoretical calculations section) we find a rate constant kr = 9.87∙1011 s−1, characterizing the reaction as an ultrafast one, with a corresponding life-time of
- factor of 1.43 [11]. Theoretical calculations Calculations for the photodegradation of carbamates The structures, properties and the basic photochemistry of compounds 11 and 12 was studied using the density functional theory (DFT) method [86][87][88][89] and the functional B3PW91 along with the 6-31 G(d













Pigmentosins from Gibellula sp. as antibiofilm agents and a new glycosylated asperfuran from Cordyceps javanica
Beilstein J. Org. Chem. 2019, 15, 2968–2981, doi:10.3762/bjoc.15.293

- File 1. ECD theoretical calculations TDDFT-ECD was used to perform theoretical ECD calculations. Conformational searches for the investigated compounds were first performed with a MMFF94S force field and an energy window of 10 kcal/mol using Omega2 software [54][55]. Each resulting conformer was then







A combinatorial approach to improving the performance of azoarene photoswitches
Beilstein J. Org. Chem. 2019, 15, 2753–2764, doi:10.3762/bjoc.15.266

- scaffold, the theoretical calculations at the PBE0-D3/6-31G** level indicate that an insertion of electron-poor fluorine atoms in the ortho-position of the aryl ring (4pzH-F1 and 4pzH-F2) leads to an increase in the half-life from ca. 1000 days in 4pzH to 2000 days in 4pzH-F1 and to 4000 days in 4pzH-F2
- above, theoretical calculations indicate that the addition of ortho-fluoro atoms leads to higher Z-isomer stability, whereas the opposite effect is found upon chlorine ortho-substitution (Table 1). The insertion of one and two electron-donating OMe and Pyr groups systematically improve the Z-isomer
- compared to E to allow for efficient Z–E photoswitching. Theoretical calculations indicate that the family of ortho-substituted 4pzH-X photoswitches provide a weak n–π* transition, with oscillator strengths f < 0.020. In contrast, 4pzMe-X derivatives show a relatively intense n–π* excitation, with f









Plasma membrane imaging with a fluorescent benzothiadiazole derivative
Beilstein J. Org. Chem. 2019, 15, 2644–2654, doi:10.3762/bjoc.15.257

- bioimaging experiments and proved to be stable under constant light irradiation for more than 4 hours (see Figure S4 in Supporting Information File 1). Theoretical calculations were then performed for a better comprehension of the photophysical data obtained for BTD-4APTEG by means of the time-dependent
- affinity for the plasma membrane as depicted in the imaging (qualitative and quantitative) experiments. Theoretical calculations were found to be in accordance with the experimental data and helped to understand the ICT stabilizing process of the designed fluorophore. The developed green emitter was
- ) 156.4, 155.8, 144.1, 130.9, 129.2, 126.4, 123.3, 120.1, 111.9. 71.50, 71.47, 70.2, 69.9, 69.3, 38.9; HRMS (ESI-Q-TOF) calcd. for C18H23N4O3S+, 375.1485; found, 375.1460. Theoretical calculations. All DFT calculations were performed using the Gaussian 09 suite of programs [64]. Geometry optimizations









AgNTf2-catalyzed formal [3 + 2] cycloaddition of ynamides with unprotected isoxazol-5-amines: efficient access to functionalized 5-amino-1H-pyrrole-3-carboxamide derivatives
Beilstein J. Org. Chem. 2019, 15, 2623–2630, doi:10.3762/bjoc.15.255

- carbene pathway presumed by mechanistic studies and theoretical calculations. Following our ongoing interest in the alkyne chemistry [36][37][38], we recently envisaged that the reaction of ynamides with isoxazoles could proceed under silver catalysis conditions, involving the generation of α-imino silver











Anion-driven encapsulation of cationic guests inside pyridine[4]arene dimers
Beilstein J. Org. Chem. 2019, 15, 2486–2492, doi:10.3762/bjoc.15.241

- the pyridine[4]arene dimer, and all mass spectrometric data and theoretical calculations show undoubtedly that the cation is located inside the cavity of the dimer in the gas phase. It is an interesting fact, that the binding properties of pyridine[4]arene differ from earlier reports [7][8]. However





Current understanding and biotechnological application of the bacterial diterpene synthase CotB2
Beilstein J. Org. Chem. 2019, 15, 2355–2368, doi:10.3762/bjoc.15.228

- followed by a 1,5-hydride shift. The sequential 1,2-hydride shift (C to E) route was initially suggested by theoretical calculations, which supported the overall mechanism [33][34], and this was verified via isotope labeling [34]. Such series of two 1,2-hydride shifts have previously been demonstrated
- in terpene biosynthesis. The enzyme could be understood as passive catalyst, essentially chaperoning the intermediates during the reaction cascade. It is clear that much of terpene biosynthesis can be understood by this concept. “Inherent reactivity” largely relies on theoretical calculations











Fluorinated azobenzenes as supramolecular halogen-bonding building blocks
Beilstein J. Org. Chem. 2019, 15, 2013–2019, doi:10.3762/bjoc.15.197
- boxes [35]. Therefore, we herein present a comprehensive investigation of the photochemistry of highly fluorinated azobenzenes. Our efforts are supported by theoretical calculations, showing that these azobenzenes are suitable for the use as building blocks in supramolecular architectures. Results and





Tautomerism as primary signaling mechanism in metal sensing: the case of amide group
Beilstein J. Org. Chem. 2019, 15, 1898–1906, doi:10.3762/bjoc.15.185

- , based on 4-(phenyldiazenyl)naphthalen-1-ol. According to the theoretical calculations the enol form stabilization could be achieved through a strong intramolecular hydrogen bond formed between the tautomeric hydroxy group and the carbonyl group from the tautomeric backbone. However, intermolecular
- still not appear even after 1024 scans. Theoretical calculations Quantum-chemical calculations were performed using the Gaussian 09 D.01 program suite [17]. The M06-2X functional [18][19] was used with the 6-31++G** basis set for the calculations. This fitted hybrid meta-GGA functional with 54% HF









Synthesis, photophysical and electrochemical properties of pyridine, pyrazine and triazine-based (D–π–)2A fluorescent dyes
Beilstein J. Org. Chem. 2019, 15, 1712–1721, doi:10.3762/bjoc.15.167

- due to the increase in the electron-withdrawing ability of the azine ring in the order of pyridyl < pyrazyl < triazyl, resulting in a decrease in the energy gap between the HOMO and the LUMO. Theoretical calculations In order to examine the HOMO and LUMO distributions of OUY-2, OUK-2 and OUJ-2, the








Superelectrophilic carbocations: preparation and reactions of a substrate with six ionizable groups
Beilstein J. Org. Chem. 2019, 15, 1515–1520, doi:10.3762/bjoc.15.153

- electrophiles based on the triarylmethyl cation scaffold (3–5, Scheme 1) [11][12]. These systems utilized pyridyl rings to produce increasing amounts of positive charge adjacent to the carbocation center. Both theoretical calculations and experimental results indicated that the carbocation center undergoes a
- position and deprotonation of the para-carbon to complete the arylation step. For the cyclization product 11, theoretical calculations indicate that cyclization requires deprotonation at the pyridinium ring [11]. Thus, either the tetracation 15 or the pentacation 16 is the likely precursor to the pyrido








Selenophene-containing heterotriacenes by a C–Se coupling/cyclization reaction
Beilstein J. Org. Chem. 2019, 15, 1379–1393, doi:10.3762/bjoc.15.138

- (Figure 2b), because a completely flat geometry of the isolated molecule DST 3 (in the gas phase) was obtained from theoretical calculations (vide infra). Molecules of DST 3 order in a typical herringbone fashion, where the terminal hydrogen atoms form hydrogen bond-like C–H heteroatom interactions (2.819
- the crystal structure analysis. The analysis of the theoretical calculations gave also insight into the electronic properties of the heterotriacene series. The energies of the calculated frontier orbitals and electronic transitions are summarized in Table 3. In this respect, the energy of the HOMO
- to the planar π-conjugated system. Gaussian deconvolution of the experimental spectra exemplarily shown for DDT 1 (Figure 6, right) evidenced the coexistence of two electronic transitions under the absorption curve in correlation with the theoretical calculations (vide infra). The absorption maxima











