Search results
Search for "inhibitor" in Full Text gives 432 result(s) in Beilstein Journal of Organic Chemistry. Showing first 200.
Bipolenins K–N: New sesquiterpenoids from the fungal plant pathogen Bipolaris sorokiniana
Beilstein J. Org. Chem. 2019, 15, 2020–2028, doi:10.3762/bjoc.15.198

- effects on cells infected with respiratory syncytial virus [22][23], induction of aerial mycelium formation in Fusarium culmorum [24], and as an inhibitor of ubiquinol-cytochrome c reductase binding protein, blocking mitochondrial ROS-mediated vascular endothelial growth factor receptor type 2 signalling






A review of the total syntheses of triptolide
Beilstein J. Org. Chem. 2019, 15, 1984–1995, doi:10.3762/bjoc.15.194

- pathways involved in the regulation of reactive oxygen species (ROS) and/or nitric oxide (NO) [9], histone methyltransferase [10], HSP70 [11], Jak2, Bcl-2/Bax [12], caspase 8 [13], NF-κB [14], X-linked inhibitor of apoptosis protein (XIAP) [15], MAPK, PI3K [16], and MPK1, ERK-1/2, and JNK-1/2 [17]. The











Archangelolide: A sesquiterpene lactone with immunobiological potential from Laserpitium archangelica
Beilstein J. Org. Chem. 2019, 15, 1933–1944, doi:10.3762/bjoc.15.189
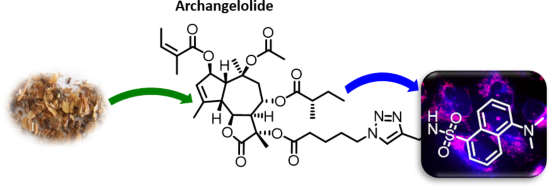
- evokes the synthesis of IL-6, INF-γ and TNF-α (see section “Abbreviations” at the end of the text). Furthermore, compound 2 has a very similar structure to the well-described SL thapsigargin, which is the best-known inhibitor of sarcoplasmic/endoplasmic reticular calcium ATPase (SERCA) with Ki values in
- reticulum and mitochondrial markers were performed. As it is apparent from Figure 3 and Figure S20 (Supporting Information File 1), compound 5 localized in the endoplasmic reticulum, which is in agreement with the site of localization of another SL, the SERCA inhibitor compound 2 [8]. However, we observed
- that this SL does not act as SERCA inhibitor. Therefore, we proceeded to confirm this hypothesis by a molecular dynamics simulation study. Molecular dynamics simulation of compounds 1 and 2 with SERCA The binding cavity for thapsigargin and compound 2 in the SERCA protein lies in the transmembrane












Installation of -SO2F groups onto primary amides
Beilstein J. Org. Chem. 2019, 15, 1907–1912, doi:10.3762/bjoc.15.186

- binding sites (Figure 1) [20]. The smallest member of this family, methyl sulfonyl fluoride (MSF), is known as a selective and irreversible inhibitor of acetylcholinesterase (AChE) [21][22]. The sulfonyl fluoride inhibitors NSC 127755 was found for specifically modifying tyrosine-31 of DHFR in chicken






N-(1-Phenylethyl)aziridine-2-carboxylate esters in the synthesis of biologically relevant compounds
Beilstein J. Org. Chem. 2019, 15, 1722–1757, doi:10.3762/bjoc.15.168

- )-7 (Scheme 6) which was found to be an effective inhibitor of the mitotic kinesin. The biologically active enantiomer of mexiletine (R)-24 was efficiently synthesized from the alcohol (2R,1'R)-7 (Scheme 7) [45]. When the respective tosylate (2R,1'R)-25 was treated with 2,6-dimethylphenoxide two
- protected diamine (R)-80a. Removal of the chiral auxiliary yielded the diamine (R)-80b which after acylation provided analogues (R)-76. Compound (R)-76 (R = C9H19) appeared inactive as inhibitor of human sphingosine kinases 1 and 2. 1,2-Diamino-3-hydroxy derivatives The interest in sphingoid analogues stems
- from the involvement of sphingolipid metabolites in an array of important cell processes. ᴅ-threo-PDMP (1R,2R)-81 is a ceramide analogue identified as an inhibitor of glucosylceramide synthase (GCS) at micromolar concentrations [66][67]. It was efficiently synthesized [68][69] employing the alcohol (2R
































































Synthesis of 9-O-arylated berberines via copper-catalyzed CAr–O coupling reactions
Beilstein J. Org. Chem. 2019, 15, 1575–1580, doi:10.3762/bjoc.15.161

- elaborate motifs such as heterocycles [10][11][12], glucose [13], nitric oxide [14], and the multidrug-resistance pump inhibitor 5-nitro-2-phenylindole (INF55) [15] have been covalently attached to BBR, further widening its pharmacological spectrum. Aromatic rings are another family of substituents having





Synthesis and biological evaluation of truncated derivatives of abyssomicin C as antibacterial agents
Beilstein J. Org. Chem. 2019, 15, 1468–1474, doi:10.3762/bjoc.15.147

- covalent inhibitor of 4-amino-4-deoxychorismate (ADC) synthase, which is the enzyme that catalyzes the conversion of chorismate and glutamine into ADC and glutamate, the first step in the biosynthesis of p-aminobenzoic acid (PABA) in bacteria [6]. Specifically, AbC binds via a Michael addition between a







Synthesis of non-racemic 4-nitro-2-sulfonylbutan-1-ones via Ni(II)-catalyzed asymmetric Michael reaction of β-ketosulfones
Beilstein J. Org. Chem. 2019, 15, 1289–1297, doi:10.3762/bjoc.15.127

- frequently used in the synthesis of organic fine chemicals and natural compounds. In addition to using the sulfonyl group as an auxiliary, it is also included in some chiral bioactive molecules, such as remikiren (1, renin inhibitor for the treatment of hypertension) [5][6], eletriptan (2, Relpax®, serotonin
- 5-HT1 receptor agonist for the treatment of migraine) [7], and apremilast (3, Otezla®, inhibitor of the PDE4 for the treatment of certain types of psoriasis and psoriatic arthritis) [8] (Figure 1). Recently we have shown that racemic sulfone 4 exhibits high antiviral activity against BVDV with low








Phylogenomic analyses and distribution of terpene synthases among Streptomyces
Beilstein J. Org. Chem. 2019, 15, 1181–1193, doi:10.3762/bjoc.15.115

- the recombinant enzyme from Streptomyces malaysiensis [43]. The diterpene 7 is a precursor to the lysophospholipase inhibitor cyclooctatin (20) formed by the action of two genetically clustered cytochrome P450 monooxygenases CotB3 and CotB4 (Scheme 4) [40][44], while no derivatives from 8 are










Insertion of [1.1.1]propellane into aromatic disulfides
Beilstein J. Org. Chem. 2019, 15, 1172–1180, doi:10.3762/bjoc.15.114

- ) [1]. In their pioneering work Stepan et al. replaced a para-substituted fluorophenyl ring in the γ-secretase inhibitor BMS-708,163 with a BCP whereby the oral absorption and in vitro metabolic stability could be significantly increased [2]. BCPs are usually derived from [1.1.1]propellane (1) [7










Synthesis of aryl cyclopropyl sulfides through copper-promoted S-cyclopropylation of thiophenols using cyclopropylboronic acid
Beilstein J. Org. Chem. 2019, 15, 1162–1171, doi:10.3762/bjoc.15.113

- ]. Roniciclib, also named BAY 1000394, is a pan-cyclin-dependant kinase (CDK) inhibitor that contains an aryl cyclopropyl sulfoximine and that was developed to treat patients with untreated small cell lung cancer [6][9]. Aryl cyclopropyl sulfides 1 are also remarkable synthons in organic synthesis (Scheme 1





Stereo- and regioselective hydroboration of 1-exo-methylene pyranoses: discovery of aryltriazolylmethyl C-galactopyranosides as selective galectin-1 inhibitors
Beilstein J. Org. Chem. 2019, 15, 1046–1060, doi:10.3762/bjoc.15.102

- most potent galectin-1 inhibitor 1b has at least fiftyfold better affinity than the reference monosaccharide methyl β-D-galactoside (11) and a similar affinity as the reference disaccharide methyl β-lactoside (12). Hence, the 4-fluorophenyltriazol moiety of 1b efficiently replaces the galectin-1
- -interacting glucose unit in methyl β-lactoside (12) and at the same time induces a significantly better selectivity than that of methyl β-lactoside (12). Taken together, the 4-fluorophenyltriazole 1b represents the most potent mono-galactoside-derived galectin-1 inhibitor, albeit with an apparently lower




Novel (2-amino-4-arylimidazolyl)propanoic acids and pyrrolo[1,2-c]imidazoles via the domino reactions of 2-amino-4-arylimidazoles with carbonyl and methylene active compounds
Beilstein J. Org. Chem. 2019, 15, 1032–1045, doi:10.3762/bjoc.15.101

- -dihydro analogs are represented only by several substances [33][34]. Partially hydrogenated pyrrolo[1,2-c]imidazole is a part of (±)-axinellamines 11. 4-[(5R)-6,7-Dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl]-3-fluorobenzonitrile (LCI-699, osilodrostat) is considered as an inhibitor of aldosterone synthase














Heck- and Suzuki-coupling approaches to novel hydroquinone inhibitors of calcium ATPase
Beilstein J. Org. Chem. 2019, 15, 971–975, doi:10.3762/bjoc.15.94

- Abstract In this study, we explored Heck- and Suzuki-coupling methodology to modify the template 2,5-di-tert-butylhydroquinone (BHQ, 2), an inhibitor of the enzyme sarco/endoplasmic reticulum calcium ATPase (SERCA). We found that by utilizing Suzuki coupling, we could successfully attach a six-carbon
- cytotoxic agent. The problem of concomitant toxicity to healthy cells has been circumvented by attaching a short peptide (His-Ser-Ser-Lys-Leu-Gln-Leu) to a tether at TG’s C-8 position (1b). This modification renders the inhibitor inactive [3]. Prostate cancer cells produce on their surface the serine
- inhibiting SERCA and preventing it from loading intracellular calcium stores. No other major proteases share the specificity of PSA, which prevents premature inhibitor activation in healthy cells. Moreover, as PSA is deactivated by inhibitors present in the blood serum, potential detrimental effects on other





Towards the preparation of synthetic outer membrane vesicle models with micromolar affinity to wheat germ agglutinin using a dialkyl thioglycoside
Beilstein J. Org. Chem. 2019, 15, 937–946, doi:10.3762/bjoc.15.90

- pH 7.4 containing 0.05% (v/v) Tween 20). This washing procedure was repeated after each incubation step. The coated microtiter plates were then blocked with BSA in PBS (3% w/v, 1 h at 37 °C, 100 μL per well). Serial two-fold dilutions of each inhibitor were pre-incubated 1 h at 37 °C in PBS-DMSO (9:1
- using the following equation, where A is absorbance. % Inhibition = [(A(no inhibitor) – A(with inhibitor)/A(no inhibitor)] × 100. The percent of inhibition was plotted against the logarithm of the concentration of the sugar derivatives. The sigmoidal curves were fitted and the concentration at 50








![[Graphic 2]](/bjoc/content/inline/1860-5397-15-90-i4.svg?max-width=637&scale=0.472728) ) and (○) ...
) and (○) ...

Synthesis of (macro)heterocycles by consecutive/repetitive isocyanide-based multicomponent reactions
Beilstein J. Org. Chem. 2019, 15, 906–930, doi:10.3762/bjoc.15.88

- synthesis of telaprevir 64, a protease inhibitor used in the treatment of hepatitis C, through a very short and efficient synthetic strategy involving as key steps two IMCRs (Ugi and Passerini) [28]. The strategy involved the synthesis of isocyanide 58 via a Passerini reaction using aldehyde 56, cyclopropyl


































Efficient synthesis of pyrazolopyridines containing a chromane backbone through domino reaction
Beilstein J. Org. Chem. 2019, 15, 874–880, doi:10.3762/bjoc.15.85

- tracazolate, cartazolate, and etazolate [22]. Other pyrazolopyridine-containing bioactive compounds include a GSK-3 inhibitor [23] and BAY 41-2272, [24][25] and could be used as cardiovascular therapeutic agents (Figure 1). Moreover, pyrazolopyridine derivatives also have industrial importance as fluorophores







New sesquiterpenoids from the South China Sea soft corals Clavularia viridis and Lemnalia flava
Beilstein J. Org. Chem. 2019, 15, 695–702, doi:10.3762/bjoc.15.64

- PTP1B inhibitory assay, the inhibitory effects of compounds 1–8 were evaluated against PTP1B, and the result showed that compounds 1, 2 and 4 had a moderate PTP1B inhibitory activity with IC50 values of 18.8, 21.8 and 15.6 μM, respectively. The known PTP1B inhibitor oleanolic acid (IC50 = 3.0 μM) were
- inhibitor concentration [I] by using the following equation: % inhibition = 1/(1 + [IC50/[I]]k), where k is the Hill coefficient; IC50 ≥ 50 μM was considered inactive. NF-κB signaling pathway inhibitory activity assays NF-κB signaling pathway inhibitory activity was evaluated according to the previously
- 4 software (Graphpad, San Diego, CA) from the nonlinear curve fitting of the percentage of inhibition (% inhibition) versus the inhibitor concentration [I] by using the following equation: % inhibition = 100/(1 + [IC50/[I]]k), where k is the Hill coefficient. Bortezomib was used as a positive




Cyclopropene derivatives of aminosugars for metabolic glycoengineering
Beilstein J. Org. Chem. 2019, 15, 584–601, doi:10.3762/bjoc.15.54














Synthesis and SAR of the antistaphylococcal natural product nematophin from Xenorhabdus nematophila
Beilstein J. Org. Chem. 2019, 15, 535–541, doi:10.3762/bjoc.15.47

- described as a potent inhibitor of various proteases, in particular trypsin and thrombin [17][18][19]. Hereby, the α-keto amide covalently binds to the serine oxygen in the active site under formation of a stable tetrahedral hemiketal. Furthermore, substitution of the indole hydrogen by alkyl, aryl or




Design of indole- and MCR-based macrocycles as p53-MDM2 antagonists
Beilstein J. Org. Chem. 2019, 15, 513–520, doi:10.3762/bjoc.15.45

- most of the human cancers have either mutated the p53 itself or the p53 pathway is inhibited. The latter group of tumors retains the wild type p53 (wt-p53) but its pathway is inactivated by negative regulators, mainly the MDM2 and MDMX proteins. Thus, the design and synthesis of an inhibitor of the
- Leu57. This pocket when filled with a smaller hydrophobic substituent such as -Cl boosts the inhibitor activity in accordance with literature [33]. Conclusion We effectively synthesized p53-MDM2 antagonists based on an artificial macrocyclic scaffold. 16 different derivatives were obtained and screened
- probing the subpockets of MDM2 and expansion of the chemistry compared to previous studies [13]. (A) Overlay of 1 H,15N-HSQC spectra of the reference MDM2 (red) and the titration steps with the 2i inhibitor. MDM2/2i ratios 4:1 (orange), 4:2 (yellow), 4:3 (green), 1:1 (light blue), 1:2 (blue), 1:5 (purple






A chemoenzymatic synthesis of ceramide trafficking inhibitor HPA-12
Beilstein J. Org. Chem. 2019, 15, 490–496, doi:10.3762/bjoc.15.42

- (HPA-12, 1, Figure 1) as the first inhibitor of CERT-mediated ceramide transport [11]. However, the initially determined (1R,3R) configuration of the most active HPA-12 stereoisomer (compound 1, Figure 1) was later revised to (1R,3S) configuration (compound 2, Figure 1) by Berkeš et al. in 2011 [12






Aqueous olefin metathesis: recent developments and applications
Beilstein J. Org. Chem. 2019, 15, 445–468, doi:10.3762/bjoc.15.39

- [70]. α-Chymotrypsin is a serine protease that recognizes hydrophobic residues in one of its clefts. A modified HG-type catalyst (66) contains an L-phenyl chloromethyl ketone moiety that acts as inhibitor and is first recognized by supramolecular anchoring and then covalently attaches upon
- , entries 2, 7 and 12). Gebbink and co-workers anchored the HG-type catalyst 79 to cutinase, a serine hydrolase [75]. The phosphonate ester moiety acts as a suicide inhibitor forming an irreversible covalent bond to a serine residue present in the active site of the enzyme. Assembly of ArM 8 occurs at pH 5



























Study on the regioselectivity of the N-ethylation reaction of N-benzyl-4-oxo-1,4-dihydroquinoline-3-carboxamide
Beilstein J. Org. Chem. 2019, 15, 388–400, doi:10.3762/bjoc.15.35

- eigenvalue in the Hessian second order matrix) [28][38][39][40][41]. Structures of some bioactive 4-oxoquinoline-3-carboxamide derivatives 1–4 with different bioactive profiles. Ki = binding affinity; AHA = acetohydroxamic acid (standard urease inhibitor); SAHA = suberoylanilide hydroxamic acid (an FDA












Synthesis and biological activity of methylated derivatives of the Pseudomonas metabolites HHQ, HQNO and PQS
Beilstein J. Org. Chem. 2019, 15, 187–193, doi:10.3762/bjoc.15.18

- metabolites [16]. For example, Mycobacterium abscessus, which like P. aeruginosa and S. aureus can occur in the lung of cystic fibrosis patients [17][18][19], is able to methylate HQNO to give 2-heptyl-1-methoxy-4(1H)-quinolone (HMOQ). HMOQ is a significantly less efficient inhibitor of the respiratory chain






