Search results
Search for "simulations" in Full Text gives 139 result(s) in Beilstein Journal of Organic Chemistry.
How and why plants and human N-glycans are different: Insight from molecular dynamics into the “glycoblocks” architecture of complex carbohydrates
Beilstein J. Org. Chem. 2020, 16, 2046–2056, doi:10.3762/bjoc.16.171

- undetected because of their intrinsic structural disorder. In this work we use molecular dynamics (MD) simulations to provide insight into N-glycans’ 3D structure by analysing the effects of a set of very specific modifications found in plants and invertebrate N-glycans, which are immunogenic in humans. We
- represents an essential resource to advance glycomics [11][12][13][14][15], while molecular simulations fit in very well as complementary and orthogonal techniques to support and advance structural glycobiology research. Indeed, current high performance computing (HPC) technology allows us to study realistic
- model systems [16][17] and to reach experimental timescales [18], so that computing can now contribute as one of the leading research methods in structural glycobiology. One of the most interesting and remarkably challenging areas in glycoscience research that HPC simulations can address is the study of








pH- and concentration-dependent supramolecular self-assembly of a naturally occurring octapeptide
Beilstein J. Org. Chem. 2020, 16, 2017–2025, doi:10.3762/bjoc.16.168

- (Glu−) amino acids [73][74]. Mechanistic insights into the observed conformational transformation via molecular dynamics simulations are underway in our laboratory. Conclusion In summary, we synthesized a naturally occurring amphiphilic peptide fragment, PEP-1, from a β-sheet lectin protein, galectin-1






Synthesis, docking study and biological evaluation of ᴅ-fructofuranosyl and ᴅ-tagatofuranosyl sulfones as potential inhibitors of the mycobacterial galactan synthesis targeting the galactofuranosyltransferase GlfT2
Beilstein J. Org. Chem. 2020, 16, 1853–1862, doi:10.3762/bjoc.16.152

- mechanism studies using computational chemistry methods. The probable reaction mechanisms were studied by hybrid DFT QM/MM molecular dynamics simulations [11] where the possible transition state (TS) structures were localized. The observation of the possible TS structure opens the opportunities for the in
- structures. All fructofuranose and tagatofuranose derivatives were docked into the GlfT2 structure obtained by QM/MM molecular dynamics simulations, which representes the structure close to the transition state structure of the GlfT2 catalytic reaction. The best ten docking poses of each docked molecule were










Models of necessity
Beilstein J. Org. Chem. 2020, 16, 1649–1661, doi:10.3762/bjoc.16.137

- layer of imperfection. Biological systems are even more complex, with an extraordinarily large concentration of molecules in, for instance, cells. In such systems, the necessity to sample all possible geometrical configurations of the system becomes the second major hurdle to accurate simulations. Even
- intelligence. The consequence is that we must distinguish far more clearly between the chemical ontology of AI and ML and our everyday language. The use of the Lewis model goes even further. Biomolecular simulations are based on molecular force fields, which are simply Lewis structures translated into a
- -atomic representation [73]. These force fields have attained a remarkable level of accuracy for proteins, so that force-field based simulations have become predictive in many fields of biology, medicinal chemistry and biophysics [74]. Models, approximations and paradigms Despite the many advances made





Synthesis and properties of tetrathiafulvalenes bearing 6-aryl-1,4-dithiafulvenes
Beilstein J. Org. Chem. 2020, 16, 974–981, doi:10.3762/bjoc.16.86

- bearing 1,3-dithiol-2-ylidenes took place efficiently to produce the corresponding π-conjugated molecules. We also succeeded in the estimation of the oxidation potentials and number of electrons involved in each oxidation step of the obtained compounds by digital simulations. Keywords: cross-conjugated








Design and synthesis of diazine-based panobinostat analogues for HDAC8 inhibition
Beilstein J. Org. Chem. 2020, 16, 628–637, doi:10.3762/bjoc.16.59

- the receptor using the Surflex-Dock Geom (SFXC) protocol [41][42][43] to evaluate the binding affinity of the ligand for the HDAC8 receptor. The C-scoring method was used to calculate these binding affinities and binding scores are given in −log10(Kd) values [45]. Docking simulations where ran








Two antibacterial and PPARα/γ-agonistic unsaturated keto fatty acids from a coral-associated actinomycete of the genus Micrococcus
Beilstein J. Org. Chem. 2020, 16, 297–304, doi:10.3762/bjoc.16.29

- (δC 33.1 for 2 vs 28.3 for 1), a decisive evidence was acquired from spin system simulations using the software ‘nmrpeak’ [19], which gave the best match to the experimentally obtained 1H NMR spectrum with the setting of 3JH8,H9 = 15.6 Hz and 3JH7,H8 = 9.5 Hz (Figure 3). Thus, the C8 geometry was
- system simulations were performed using the freeware nmrpeak.exe [19]. Antimicrobial assay Antimicrobial assays were carried out in a similar manner as described in [18]. The antimicrobial activity was evaluated by the liquid microculture method using round-bottomed 96-well microtiter plates against five






Unexpected one-pot formation of the 1H-6a,8a-epiminotricyclopenta[a,c,e][8]annulene system from cyclopentanone, ammonia and dimethyl fumarate. Synthesis of highly strained polycyclic nitroxide and EPR study
Beilstein J. Org. Chem. 2019, 15, 2664–2670, doi:10.3762/bjoc.15.259

- molecules along the a-axis. EPR measurements Figure 4 demonstrates the X-band CW EPR spectra of nitroxide 6 in water/glycerol solution at 180 K and at room temperature with simulations (red) using the parameters listed in the caption. The electron spin relaxation of nitroxides with different bulky
- experimental spectra and the red lines are simulations for hfi, g-factor and correlation times of A(N) = [0.71; 0.71; 3.71] mT, g = [2.0087; 2.0055; 2.0019], tcorr = 0.53 ns (at 298 K). Synthesis of compound 1. Possible mechanism for the formation of 1. Synthesis of nitroxide 6. A proposed mechanism for










Current understanding and biotechnological application of the bacterial diterpene synthase CotB2
Beilstein J. Org. Chem. 2019, 15, 2355–2368, doi:10.3762/bjoc.15.228

- date, CotB2 represents the best studied bacterial diterpene synthase. Its reaction mechanism has been addressed by isoptope labeling, targeted mutagenesis and theoretical computations in the gas phase, as well as full enzyme molecular dynamic simulations. By X-ray crystallography different snapshots of
- ], density functional theory calculations [33] as well as QM/MM simulations [37]. Variants of CotB2 open the route to a novel product portfolio with altered cyclic carbon skeletons, which can be converted into bioactive compounds by chemo-enzymatic methodologies. Modification descriptions are composed











α-Photooxygenation of chiral aldehydes with singlet oxygen
Beilstein J. Org. Chem. 2019, 15, 2076–2084, doi:10.3762/bjoc.15.205

- force field within 5 kcal/mol energy ranges. Further optimization was carried out at DFT level using the B3LYP functional and the Def2TZVP basis set in the Gaussian 09 [36]. Simulations of ECD spectra were carried out with TD-DFT methods for conformers found in the range of 2.5 kcal/mol. The B3LYP








Archangelolide: A sesquiterpene lactone with immunobiological potential from Laserpitium archangelica
Beilstein J. Org. Chem. 2019, 15, 1933–1944, doi:10.3762/bjoc.15.189
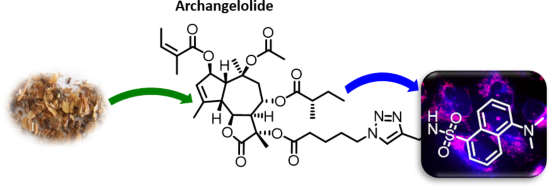
- ). The first round of simulations (1–10 ns) of compound 1 docking into SERCA cavity with compound 1 constrained as described (simulation 1) showed the presence of hydrophobic interactions of ligand side chains with Phe256, Val263, Ile829 and the hydrophobic part of Gln259 amino acid residues. Gln259 was
- the main residue involved in hydrophilic interaction. Interestingly, during the simulations in water environment, compound 1 had the tendency to escape from the SERCA cavity. In simulation 2 of compound 1 rotated by 180° with constrained positions (10 ns), interactions with Phe256 and hydrophobic
- and Figure 5). During simulations with SERCA positioned in a phospholipid membrane (simulation 4; 1 ns), the free space between individual phospholipids in the cell membrane was filled. SERCA remained stable and tightly embedded in the membrane and did not show any tendency to escape from it. Compound












Functional panchromatic BODIPY dyes with near-infrared absorption: design, synthesis, characterization and use in dye-sensitized solar cells
Beilstein J. Org. Chem. 2019, 15, 1758–1768, doi:10.3762/bjoc.15.169

- also a reduced energy driving force for charge separation at the TiO2 surface. To check this hypothesis, we performed TD-DFT simulations of the optical absorption spectra of the 4 representative dyes. The results reported in Figure 3 are fully consistent with the predictions from the one-electron








Water inside β-cyclodextrin cavity: amount, stability and mechanism of binding
Beilstein J. Org. Chem. 2019, 15, 1592–1600, doi:10.3762/bjoc.15.163

- disordered and mobile, and that the OH groups of the host β-CD may rotate [17]. Studies with molecular dynamics simulations have found only four water molecules inside the host β-CD cavity [18]. The energetics of the CD hydration/dehydration have been investigated as well. Experimental studies have been able







Synthesis and conformational preferences of short analogues of antifreeze glycopeptides (AFGP)
Beilstein J. Org. Chem. 2019, 15, 1581–1591, doi:10.3762/bjoc.15.162

- into the ice lattice [12], others investigated the adsorption process on different surfaces via atomic force microscopy [10][13][14]. The antifreeze activity has been correlated with long-range perturbation of hydration dynamics [15]. Latest molecular dynamics simulations suggest that AFGP reversibly






Transient and intermediate carbocations in ruthenium tetroxide oxidation of saturated rings
Beilstein J. Org. Chem. 2019, 15, 1552–1562, doi:10.3762/bjoc.15.158

- , the proposed asynchronous concerted mechanism. A deeper analysis of the full path of the reaction using MD calculations [29] would be needed in order to assess the synchronicity and life time of transient species [30]. The recent use of MD simulations has demonstrated that a single transition state
- some chemical behavior. Indeed, further theoretical studies on MD simulations would be needed to elucidate the lifetime of the transient carbocation [34][35]. These results demonstrate the one-step-two-stage character [42] of the ruthenium oxidations of alkanes in which H transfer and O–C bond












Host–guest interactions between p-sulfonatocalix[4]arene and p-sulfonatothiacalix[4]arene and group IA, IIA and f-block metal cations: a DFT/SMD study
Beilstein J. Org. Chem. 2019, 15, 1321–1330, doi:10.3762/bjoc.15.131

- anchoring points for the positively charged guests. Cation–π interactions between the monoatomic cations and p-sulfonatocalix[4]arene in water are supposed (but not proven) to take part in the inclusion complex formation [31]. Mendes et al. have carried out molecular dynamics (MD) simulations of association
- upon binding has been noted [31]. A model of hydrated, by an average number of 10 water molecules, La3+ cation has already been used by Mendes et al. in the MD simulations of p-sulfonatocalix[4]arene association with rare-earth metal cations and organic cations in aqueous solutions [32]. The effect of








Stereo- and regioselective hydroboration of 1-exo-methylene pyranoses: discovery of aryltriazolylmethyl C-galactopyranosides as selective galectin-1 inhibitors
Beilstein J. Org. Chem. 2019, 15, 1046–1060, doi:10.3762/bjoc.15.102

- galectin-1 (pdb id 1GZW) and galectin-3 (pdb id 1KJL), respectively, and with the 4-fluorophenyltriazol ring protruding away from the protein. The simulations with 1b and galectin-1 converged toward a complex geometry in which the 4-fluorophenyltriazole extended along a shallow groove formed by Trp68-Gly69
- the weaker affinity by the corresponding 2-fluorophenyltriazole analogue 1d, because in the favored complex geometry of 1b introduction of a 2-fluoro atom would lead to this atom being close to either the Glu71 carboxylate or the triazole N3 lone pair. MD simulations with 1b positioned in a similar




Influence of per-O-sulfation upon the conformational behaviour of common furanosides
Beilstein J. Org. Chem. 2019, 15, 685–694, doi:10.3762/bjoc.15.63

- overlapping signals J coupling constants were extracted from 2nd order spectra simulations using Bruker TopSpin software (DAISY). The obtained results (see Tables 1–3) showed good coincidence with previously published data for related monosaccharides [15][26][27]. As can be seen from Table 3, J coupling





Polyaminoazide mixtures for the synthesis of pH-responsive calixarene nanosponges
Beilstein J. Org. Chem. 2019, 15, 633–641, doi:10.3762/bjoc.15.59

- the 2:1:1 ratio expected on the grounds of numerical simulations. The separation of such a mixture would be unsuitable in view of a possible large-scale synthesis and application of the relevant nanosponge polymeric materials, because it should involve undesirable waste of time and materials. However











Olefin metathesis in multiblock copolymer synthesis
Beilstein J. Org. Chem. 2019, 15, 218–235, doi:10.3762/bjoc.15.21

- chains hinders their ordering [19] so that the only interesting are regular multiblock copolymers that can form structures with more than one periodicity [20]. Meanwhile, theoretical investigations [21][22][23] and computer simulations [24][25][26][27] gradually revealed the high potential of random



















Adhesion, forces and the stability of interfaces
Beilstein J. Org. Chem. 2019, 15, 106–129, doi:10.3762/bjoc.15.12











Synthesis, biophysical properties, and RNase H activity of 6’-difluoro[4.3.0]bicyclo-DNA
Beilstein J. Org. Chem. 2019, 15, 79–88, doi:10.3762/bjoc.15.9

- as the 6’F-bc4,3-DNA. Furthermore, duplexes of a 6’F-bc4,3-modified strand paired to RNA unveiled in the MD simulations a flexible minor groove distance [37]. This flexibility is thought to play a crucial role for the fitting of the duplex into the DNA-binding channel and the phosphate-binding pocket
- of the enzyme. Furthermore, the phosphate-binding pocket requires a large distortion of the backbone angle α in order that the phosphate group of the AON can be positioned in it [50][51]. The 6’F-bc4,3-DNA containing strand also complied with this requirement according to the MD simulations [37








6’-Fluoro[4.3.0]bicyclo nucleic acid: synthesis, biophysical properties and molecular dynamics simulations
Beilstein J. Org. Chem. 2018, 14, 3088–3097, doi:10.3762/bjoc.14.288

- modification might be a substrate for RNase H. Keywords: DNA/RNA affinity; fluorinated cyclopropanes; fluorinated nucleic acids; molecular dynamics simulations; sugar modified nucleosides; Introduction A powerful strategy for the treatment of various disorders like cancer, viral and inherited diseases is the
- unit and possibly positively impact the duplex stability. Here we report on the synthesis of the two 6’F-bc4,3 pyrimidine analogs with the base T and C, their incorporation into DNA, their biophysical properties, as well as a structural analysis by molecular dynamics simulations of hybrid DNA and RNA
- structure, giving evidence of mixed A/B-type helices. Molecular modeling To gain more information on the structural features of the 6’F-bc4,3 modification, we performed molecular dynamics simulations of the modified duplexes. We first calculated the potential energy profile versus pseudorotation phase angle









Dispersion-mediated steering of organic adsorbates on a precovered silicon surface
Beilstein J. Org. Chem. 2018, 14, 2715–2721, doi:10.3762/bjoc.14.249

- dispersion or covalent interactions. At very short distances, Pauli repulsion creates the repulsive potential wall. Experimental studies in combination with Monte Carlo simulations have shown that growth of 1 on Si(001) results in non-statistical formation of chains with an average distance of 1.5 to 2







Learning from B12 enzymes: biomimetic and bioinspired catalysts for eco-friendly organic synthesis
Beilstein J. Org. Chem. 2018, 14, 2553–2567, doi:10.3762/bjoc.14.232

- not been reported in the literature, with the exception of electrocatalytic systems [26][27]. To achieve functional simulations of B12 enzymes under non-enzymatic conditions, our strategy is to fabricate the artificial enzymes by combining a functional equivalent of B12 and that of an apoenzyme
- catalytic reactions We deeply investigated the electrochemical catalytic reactions mediated by 1 and related complexes and succeeded in the functional simulations of MMCM-type 1,2-migration reactions [42]. For example, when 2,2-bis(ethoxycarbonyl)-1-bromopropane was selected as a model substrate, the 1,2















