Search results
Search for "targeting" in Full Text gives 205 result(s) in Beilstein Journal of Organic Chemistry. Showing first 200.
Synthesis of a tubugi-1-toxin conjugate by a modulizable disulfide linker system with a neuropeptide Y analogue showing selectivity for hY1R-overexpressing tumor cells
Beilstein J. Org. Chem. 2019, 15, 96–105, doi:10.3762/bjoc.15.11

- Pharmaceuticals GmbH, Leberstr. 20, A-1110 Vienna, Austria 10.3762/bjoc.15.11 Abstract Tubugi-1 is a small cytotoxic peptide with picomolar cytotoxicity. To improve its cancer cell targeting, it was conjugated using a universal, modular disulfide derivative. This allowed conjugation to a neuropeptide-Y (NPY
- )-inspired peptide [K4(C-βA-),F7,L17,P34]-hNPY, acting as NPY Y1 receptor (hY1R)-targeting peptide, to form a tubugi-1–SS–NPY disulfide-linked conjugate. The cytotoxic impacts of the novel tubugi-1–NPY peptide–toxin conjugate, as well as of free tubugi-1, and tubugi-1 bearing the thiol spacer (liberated from
- triple-negative breast cancer MDA-MB-468 cells. Keywords: drug targeting; neuropeptide Y; PDC; peptide–drug conjugate; targeted tumor therapy; tubugi; tubulysin A; Ugi reaction; Introduction Until recently, the medication of tumor diseases was primarily based on more or less unspecific







Repurposing the anticancer drug cisplatin with the aim of developing novel Pseudomonas aeruginosa infection control agents
Beilstein J. Org. Chem. 2018, 14, 3059–3069, doi:10.3762/bjoc.14.284

- targeting important components of the immune system, such as the type III secretion systems (T3SS), which was shown to be associated with poor clinic outcomes in patients with lower respiratory infections [8] and ventilator-associated pneumonia [9]. Identifying antimicrobial compounds which actively target








Protein–protein interactions in bacteria: a promising and challenging avenue towards the discovery of new antibiotics
Beilstein J. Org. Chem. 2018, 14, 2881–2896, doi:10.3762/bjoc.14.267

- reducing the burden of multidrug-resistant microorganisms. Protein–protein interactions (PPIs) are involved in a myriad of vital cellular processes and have become an attractive target to treat diseases. Therefore, targeting PPI networks in bacteria may offer a new and unconventional point of intervention
- in fact there are focal points (i.e., hot spots or hot segments) that contribute to the majority of the binding energy [17][18]. Targeting these “druggable” sites can therefore be used for the rational design of new therapeutic compounds that can disrupt those critical interactions. However, their
- ]. Similarly to the aforementioned sliding clamp, the structural arrangement of most bacterial SSBs differs significantly to its homolog in eukaryotic cells (replication protein A, RPA) [74]. This dissimilarity could be beneficial from a drug discovery perspective because it would enable selective targeting of










Synthesis of pyrrolidine-based hamamelitannin analogues as quorum sensing inhibitors in Staphylococcus aureus
Beilstein J. Org. Chem. 2018, 14, 2822–2828, doi:10.3762/bjoc.14.260

- resistance problem need to be multifactorial. Next to disease prevention and the development of new antibiotics, it is essential to investigate innovative strategies to combat bacterial infections [3][4]. Recently, targeting bacterial virulence has gained a lot of attention [5][6][7]. It has been










Synthesis of α-D-GalpN3-(1-3)-D-GalpN3: α- and 3-O-selectivity using 3,4-diol acceptors
Beilstein J. Org. Chem. 2018, 14, 2805–2811, doi:10.3762/bjoc.14.258

- nucleophilicity and accessibility of the axial 4-OH versus the equatorial 3-OH [40], we wondered, if this could be used to simplify the synthesis of the α-D-GalpN3-(1-3)-D-GalpN3 unit. When consulting the literature, surprisingly few reports were describing diol glycosylations targeting the galacto-configured







Novel solid-phase strategy for the synthesis of ligand-targeted fluorescent-labelled chelating peptide conjugates as a theranostic tool for cancer
Beilstein J. Org. Chem. 2018, 14, 2665–2679, doi:10.3762/bjoc.14.244

- successfully designed and demonstrated a novel continuous process for assembling targeting ligands, peptidic spacers, fluorescent tags and a chelating core for the attachment of cytotoxic molecules, radiotracers, nanomaterials in a standard Fmoc solid-phase peptide synthesis in high yield and purity. The
- groups such as Trt (chlorotrityl), Mtt (4-methyltrityl), Mmt (4-methoxytrityl) are employed to synthesise the ligand targeted fluorescent tagged bioconjugates. Using this methodology, DUPA rhodamine B conjugate (DUPA = 2-[3-(1,3-dicarboxypropyl)ureido]pentanedioic acid), targeting prostate specific
- membrane antigen (PSMA) expressed on prostate, breast, bladder and brain cancers and pteroate rhodamine B, targeting folate receptor positive cancers such as ovarian, lung, endometrium as well as inflammatory diseases have been synthesized. In vitro studies using LNCaP (PSMA +ve), PC-3 (PSMA −ve, FR −ve








Non-native autoinducer analogs capable of modulating the SdiA quorum sensing receptor in Salmonella enterica serovar Typhimurium
Beilstein J. Org. Chem. 2018, 14, 2651–2664, doi:10.3762/bjoc.14.243

- compounds represent new chemical tools for exploring the role of SdiA and QS in S. Typhimurium infections, for characterizing the mechanisms by which non-native AHLs interact with LuxR-type proteins, and for developing pathways toward novel antivirulence strategies targeting SdiA. Results and Discussion
- a small decrease in maximal activity to 80% (Figure 5B). These results support the conclusion that R8 inhibits SdiA activity by targeting SdiA directly. Itc-derivative 11 showed both competitive and non-competitive inhibition of OOHL (2) in the competition assay (Figure 5C): the EC50 increased from






The design and synthesis of an antibacterial phenothiazine–siderophore conjugate
Beilstein J. Org. Chem. 2018, 14, 2646–2650, doi:10.3762/bjoc.14.242

- achieved with beta-lactam-based siderophore conjugates targeting membrane associated penicillin binding proteins (PBPs) [7]. Cefiderocol (S-649266) is a beta-lactam–siderophore conjugate currently in phase III clinical trials which demonstrates enhanced potency against Gram-negative bacteria including
- multidrug resistant (MDR) Gram-negative pathogens [8]. One hypothesis for the success of siderophore conjugates targeting PBPs, in comparison to other antibiotic targets, is that PBPs are membrane associated and it is not necessary for the siderophore conjugate to cross into the bacterial cytoplasm [7





Targeting the Pseudomonas quinolone signal quorum sensing system for the discovery of novel anti-infective pathoblockers
Beilstein J. Org. Chem. 2018, 14, 2627–2645, doi:10.3762/bjoc.14.241

- been published based on the design and optimisation of pqs targeting QSI, which is the topic of this review. The biosynthetic cascade of the pqs QS system PQS is the abbreviation for Pseudomonas quinolone signal and actually refers to the signal molecule 2-heptyl-3-hydroxyquinolin-4(1H)-one (Figure 2
- cells in biofilms with anaerobic conditions [44]. Due to these important virulence mechanisms, which are under direct or indirect control of pqs QS, targeting this master regulatory system with small molecular compounds, thereby blocking P. aeruginosa pathogenicity, is very attractive. However, this
- attachment of a cell-penetrating peptide sequence [58]. One additional class, which did show cellular activity, was based on a catechol scaffold [59]. In analogy to the successful discovery of PqsD inhibitors starting from known FabH-targeting compounds (vide supra), ligands of another enzyme with high
























Pathoblockers or antivirulence drugs as a new option for the treatment of bacterial infections
Beilstein J. Org. Chem. 2018, 14, 2607–2617, doi:10.3762/bjoc.14.239

- current trends continue. To avoid this scenario, new classes of anti-infectives must urgently be developed. Antibiotics with new modes of action are needed, but other concepts are also currently being pursued. Targeting bacterial virulence as a means of blocking pathogenicity is a promising new strategy
- pioneering review by Clatworthy et al. in 2007, entitled ‘Targeting virulence: a new paradigm for antimicrobial therapy’ [8], which has been cited approximately 800 times. In sharp contrast to traditional antibiotics that kill or impair bacterial viability, this new approach aims to disarm the pathogen
- the aryl substitution resulted in a flat SAR with only little variation in potency among the substituents analyzed [30][36][37][38][39]. Just recently, in an attempt to search for new pharmacophores, Titz et al. have reported the synthesis of the epoxyheptose derivative 11 targeting a cysteine residue







Comparative cell biological study of in vitro antitumor and antimetastatic activity on melanoma cells of GnRH-III-containing conjugates modified with short-chain fatty acids
Beilstein J. Org. Chem. 2018, 14, 2495–2509, doi:10.3762/bjoc.14.226

- -targeting by application of a GnRH analog as a carrier to deliver a covalently linked chemotherapeutic drug directly to the tumor cells. In this study our aim was (i) to analyze the effects of GnRH-drug conjugates on melanoma cell proliferation, adhesion and migration, (ii) to study the mechanisms of tumor
- cancer cell behavior and effects of targeted chemotherapeutics with small structural differences (e.g., length of the side chain in 4Lys) was also clearly suggested. Keywords: drug-targeting conjugates; gonadotropin-releasing hormone-III; holographic microscopy; impedimetry; short-chain fatty acids
- approaches to diminish this kind of cytotoxic effects on healthy tissues is the employment of drug delivery systems directed specifically to cancer cells. The chemotherapeutic drug targeting is often based on the receptors for certain peptide hormones that are preferentially expressed by cancer cells. The









Synthesis and photophysical studies of a multivalent photoreactive RuII-calix[4]arene complex bearing RGD-containing cyclopentapeptides
Beilstein J. Org. Chem. 2018, 14, 1758–1768, doi:10.3762/bjoc.14.150

- is also faced by complexes anchored on cell-penetrating peptides. In order to provide a selective cell targeting, we developed a multivalent system composed of a photoreactive ruthenium(II) complex tethered to a calix[4]arene platform bearing multiple RGD-containing cyclopentapeptides. Extensive
- , once incorporated into targeted cancer cells thanks to the multivalent platform. Keywords: anticancer drug; calixarene; cell targeting; RGD peptide; ruthenium complex; Introduction Long-living luminescent polyazaaromatic ruthenium(II) complexes are intensively studied in a biological context, in
- of the complex [38]. It should be noted that modifications of ligands to make the resulting complexes more lipophilic or the conjugation of a complex to a CPP do not provide any control on the way these complexes will be internalized by cells and prevent thus any targeting of malignant cells over









Natural and redesigned wasp venom peptides with selective antitumoral activity
Beilstein J. Org. Chem. 2018, 14, 1693–1703, doi:10.3762/bjoc.14.144

- low as 12.5 μmol L−1 for the selective targeting of MCF-7 breast cancer cells. Flow cytometry assays further revealed that treatment with wild-type (WT) peptide Dec-NH2 led to necrosis of MCF-7 cells. Additional atomic force microscopy (AFM) measurements indicated that the roughness of cancer cell
- therapies because of their potential for selectively targeting cancer cells without harm to normal counterparts [37][38][39]. Membrane phospholipids confer permeability to the cell and regulate the flux of metabolites between the extracellular environment and the intracellular content [40]. The membrane of
- cancer cells is typically negatively charged due to a higher expression of anionic molecules such as phosphatidylserines, and negatively charged glycoproteins and glycosaminoglycans [22][23]. Here, we devised a strategy to exploit the negatively charged environment of cancer cells by targeting it with






Drug targeting to decrease cardiotoxicity – determination of the cytotoxic effect of GnRH-based conjugates containing doxorubicin, daunorubicin and methotrexate on human cardiomyocytes and endothelial cells
Beilstein J. Org. Chem. 2018, 14, 1583–1594, doi:10.3762/bjoc.14.136

- Chemistry, Pázmány P. stny 1/A, Hungarian Academy of Science, Budapest, 1117, Hungary 10.3762/bjoc.14.136 Abstract Background: Cardiomyopathy induced by the chemotherapeutic agents doxorubicin and daunorubicin is a major limiting factor for their application in cancer therapy. Chemotactic drug targeting
- : cardiotoxicity; drug targeting; GnRH-conjugates; HCM; HUVEC; impedimetry; Introduction Gonadotropin-releasing hormone (GnRH) is a peptide hormone secreted by the hypothalamus, which stimulates the release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the pituitary. Thus, it represents
- . Small peptides that recognize target receptors on tumor cells might be suitable targeting moieties for this purpose. Hormone peptides, in particular, GnRH and somatostatin derivatives that possess antiproliferative effect on their own, are among the best candidates as homing peptides [10]. A.V. Schally





Design and biological characterization of novel cell-penetrating peptides preferentially targeting cell nuclei and subnuclear regions
Beilstein J. Org. Chem. 2018, 14, 1378–1388, doi:10.3762/bjoc.14.116

- ) with selective suborganelle-targeting properties. The nuclear localization sequence N50, as well as the nucleoli-targeting sequence NrTP, respectively, were fused to a shortened version of the cell-penetrating peptide sC18. We examined cellular uptake, subcellular fate and cytotoxicity of these novel
- excluded. However, initial drug delivery studies demonstrated the high versatility of these new peptides as efficient transport vectors targeting specifically nuclei and nucleoli. In future, they could be further explored as parts of newly created peptide–drug conjugates. Keywords: anticancer drugs; cell
- nuclei; cell-penetrating peptides; nucleoli; subcellular targeting; Introduction Various drugs act on targets that are located within the nucleus, the control center of the eukaryotic cell. A lipid bilayer membrane, which is perforated with nuclear pore complex structures through which the transfer of








Acyl-group specificity of AHL synthases involved in quorum-sensing in Roseobacter group bacteria
Beilstein J. Org. Chem. 2018, 14, 1309–1316, doi:10.3762/bjoc.14.112

- enzyme PgaI2 from P. inhibens, competition experiments were performed. Targeting the optimal chain length of the fully saturated substrates first, a mixture with equal molar concentrations of the substrates 10a–h was offered to the recombinant protein. GC/MS analysis of the resulting extract (Figure 2A





Novel unit B cryptophycin analogues as payloads for targeted therapy
Beilstein J. Org. Chem. 2018, 14, 1281–1286, doi:10.3762/bjoc.14.109

- glycol spacer with a terminal azide results in a complete loss of activity. Docking studies of the novel cryptophycin analogues to β-tubulin provided a rationale for the observed cytotoxicities. Keywords: cryptophycin; cytotoxic agents; novel payloads; tubulin inhibitors; tumour targeting; Introduction







Synthetic avenues towards a tetrasaccharide related to Streptococcus pneumonia of serotype 6A
Beilstein J. Org. Chem. 2018, 14, 1095–1102, doi:10.3762/bjoc.14.95

- conjugate in proper amounts for future vaccine development can only be met via chemical synthesis. Therefore, a number of syntheses targeting the SPn 6A tetrasaccharide has been reported in literature. Initial reports of a linear synthesis were made by Vliegenthart et al. in the nineties [17][18][19][20][21








An overview of recent advances in duplex DNA recognition by small molecules
Beilstein J. Org. Chem. 2018, 14, 1051–1086, doi:10.3762/bjoc.14.93
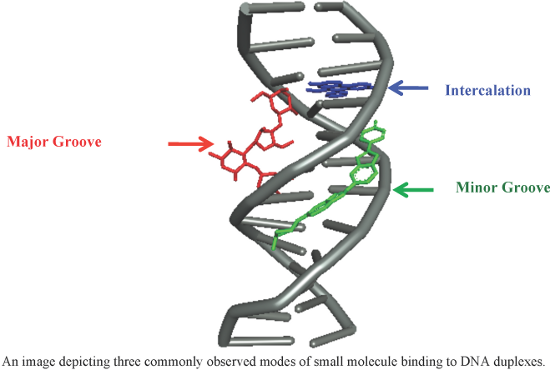
- phase synthetic approach for the syntheses of cyclic Py/Im polyamides [84]. This group previously optimized and reported a machine-assisted Fmoc solid phase synthesis of simpler polyamides to afford high step-wise coupling yield [85]. A seven-member library of cyclic polyamides targeting androgen
- response element (ARE) and the estrogen response element (ERE) was synthesized in 12–17% overall yield. Selective modifications could also be done on the GABA turn units, which showed improved cellular uptake properties. Sugiyama et al. designed and synthesized a series of telomere-targeting synthetically
- telomere foci clearly because of their fluorescent nature. Later on, the authors successfully designed tandem tetramer Py–Im polyamides with 4 hairpins and 3 hinges targeting 24 bp of the human telomere sequences [88]. Thus, the authors set the new record for the longest binding site of synthetic, non






















On the design principles of peptide–drug conjugates for targeted drug delivery to the malignant tumor site
Beilstein J. Org. Chem. 2018, 14, 930–954, doi:10.3762/bjoc.14.80

- intervention, radiation and chemotherapy. Drugs used for this purpose are inevitably cytotoxic in order to eliminate cancer cells, but they lack selectivity that could be developed through targeting malignant cells (Figure 1). Due to the uncontrolled peripheral toxicity, anticancer drugs usually kill healthy
- prodrug with enhanced plasma stability and/or cell permeability [27] and in the same time diminished toxicity for normal cells; c) covalent attachment of a drug on a tumor-targeting element (small molecule, peptide or antibody) able to selectively target and permeate cancer cells. The conjugation is being
- conducted via a rationally designed linker able to release the drug inside the cancer microenvironment [19]. The ideal targeting molecular device would consist of the following modules: a) the cytotoxic agent (drug), b) the transporting - drug delivery vehicle (i.e., lipid, mannan [28][29][30]), c) the
















Development of novel cyclic NGR peptide–daunomycin conjugates with dual targeting property
Beilstein J. Org. Chem. 2018, 14, 911–918, doi:10.3762/bjoc.14.78

- selectivity to CD13+ cells. However, the cellular uptake and cytotoxic effect of Dau=Aoa-GFLGK(c[NleNGRE]-GG-)-NH2 was higher in comparison to the control especially on HT-29 cells. Therefore, this conjugate is more suitable for drug targeting with dual targeting property. Keywords: antitumor activity; drug
- [NleNGRE]-GG)-NH2 conjugate is more suitable for drug targeting with dual acting propensity than our control lead compound. Experimental Synthesis of the novel peptide–drug conjugates Linear precursor peptides were prepared on Rink-Amide MBHA resin by SPPS. Standard Fmoc protected amino acids (Iris Biotech



Synthesis and in vitro biochemical evaluation of oxime bond-linked daunorubicin–GnRH-III conjugates developed for targeted drug delivery
Beilstein J. Org. Chem. 2018, 14, 756–771, doi:10.3762/bjoc.14.64

- Debrecen, 4032 Debrecen, Hungary 10.3762/bjoc.14.64 Abstract Gonadotropin releasing hormone-III (GnRH-III), a native isoform of the human GnRH isolated from sea lamprey, specifically binds to GnRH receptors on cancer cells enabling its application as targeting moieties for anticancer drugs. Recently, we
- cellular uptake of the bioconjugates were strongly affected by the amino acid exchange which in turn had an impact on the antitumor activity of the bioconjugates. Keywords: cytostatic effect; daunorubicin; drug-targeting; GnRH derivatives; oxime linkage; Introduction Cancer is one of the most serious
- receptors (GnRH-R) [1]. Therefore, these peptides are suitable for specific drug targeting to tumor cells. The native ligand of this receptor is GnRH-I (








Investigations towards the stereoselective organocatalyzed Michael addition of dimethyl malonate to a racemic nitroalkene: possible route to the 4-methylpregabalin core structure
Beilstein J. Org. Chem. 2018, 14, 553–559, doi:10.3762/bjoc.14.42

- stereoselective synthesis of bioactive compounds [1][2][3]. In particular, stereoselective conversions of nitro compounds served in many syntheses of pharmaceuticals [4]. Derivatives of γ-aminobutyric acid are an important class of medicines targeting problems with the central nervous system, such as pains







Stimuli-responsive oligonucleotides in prodrug-based approaches for gene silencing
Beilstein J. Org. Chem. 2018, 14, 436–469, doi:10.3762/bjoc.14.32

- enzymolabile phosphotriester groups, namely, t-Bu-SATE, OH-SATE and a conjugable aldehyde A-SATE for conjugation to delivery and targeting domains, have been selected for complete evaluation (Scheme 9A, 9B, and 9C, respectively). The optimum phosphotriester placement and number of phosphotriester groups were
- noncytotoxic fashion. Next, the authors prepared conjugates of the siRNNs via one A-SATE phosphotriester with a hepatocyte-specific tris-N-acetylgalactosamine targeting domain and demonstrated a stronger RNAi response in mouse liver (following subcutaneous or intravenous administration) than the same
- targeting the nuclease resistance of the ON prodrug. On the other hand, the thermosensitive groups are more suitable for the protection of the thiophosphates flanking the CpG motif of DNA prodrugs to provide both lipophilicity (better cellular uptake) and hydrophilicity (better solubility once groups are









































Synthesis and biological evaluation of RGD and isoDGR peptidomimetic-α-amanitin conjugates for tumor-targeting
Beilstein J. Org. Chem. 2018, 14, 407–415, doi:10.3762/bjoc.14.29

- , cyclo[DKP-RGD]-Val-Ala-PTX 5 and cyclo[DKP-isoDGR]-Val-Ala-PTX 6). Their tumor targeting ability was assessed in vitro in antiproliferative assays comparing an αVβ3 positive with an αVβ3 negative cell line [29][30]. The cyclo[DKP-isoDGR]-Val-Ala-PTX conjugate 6 displayed a remarkable targeting index (TI
- antibody α-amanitin conjugates for which the increase of cytotoxicity is 100–100000 times [5]. Therefore, despite the remarkable progresses that have been realized in recent years, integrin targeting SMDCs are still far from the clinic. Experimental Functionalized ligands H2NCH2-cyclo[DKP-RGD] (2) and





