Search results
Search for "DFT" in Full Text gives 531 result(s) in Beilstein Journal of Organic Chemistry. Showing first 200.
Reversible switching of arylazopyrazole within a metal–organic cage
Beilstein J. Org. Chem. 2019, 15, 2398–2407, doi:10.3762/bjoc.15.232

- switching to the Z isomer was accompanied by the release of one of the two guests from the cage and the formation of a 1:1 cage/Z-arylazopyrazole inclusion complex. DFT calculations suggest that this process involves a dramatic change in the conformation of the cage. Back-isomerization was induced with
- -ray diffraction. To obtain hints about the packing of Z-1 within 2, we therefore performed density functional theory (DFT) calculations. Compared with the complex before photoisomerization, the cage in (Z-1)2 is severely distorted (Figure 5; see also Supporting Information File 3), assuming a bowl
- 1:1 complexes of 2 with the Z isomers of various azobenzenes are notoriously difficult to crystallize [18]. We believe that the insights by DFT calculations reported here for (Z-1)2 can be extended to the complexes of 2 with other Z-azo guests as well. To gain further insight into the



![[Graphic 1]](/bjoc/content/inline/1860-5397-15-232-i3.svg?max-width=637&scale=1.18182) 2 (500 MHz, D2O, 298 K).
2 (500 MHz, D2O, 298 K).







Current understanding and biotechnological application of the bacterial diterpene synthase CotB2
Beilstein J. Org. Chem. 2019, 15, 2355–2368, doi:10.3762/bjoc.15.228

- rearrangement. Hong and Tantillo performed the first theoretical study of the reaction mechanism in CotB2 via gas phase density functional theory (DFT) calculations [33]. Simultaneously, Sato et al. also studied the CotB2 reaction mechanism in the gas phase using DFT, combined with experimental deuterium











Synthesis of a dihalogenated pyridinyl silicon rhodamine for mitochondrial imaging by a halogen dance rearrangement
Beilstein J. Org. Chem. 2019, 15, 2333–2343, doi:10.3762/bjoc.15.226

- microscopy [2][3][4][5][6][7][8][9], as direct probes for various biomolecules [10][11][12][13] or as sensors for metal ions [14][15][16][17][18], pH [16], voltage [19] or metabolites [20][21][22][23]. Several attempts were made, partially supported by DFT calculations, to correlate the dyes’ structural
- experiments (supported by DFT calculations) on the orbital effects of both halogens and the nitrogen position in the pyridine ring are needed to explain these effects with confidence. In addition, our SiR dye 15 displays photophysical properties (extinction coefficient, quantum yield, lifetime) in the same







Small anion-assisted electrochemical potential splitting in a new series of bistriarylamine derivatives: organic mixed valency across a urea bridge and zwitterionization
Beilstein J. Org. Chem. 2019, 15, 2277–2286, doi:10.3762/bjoc.15.220

- with triphosgene. An unsymmetrical reference urea having a NAr3 moiety, Ph1b, was also synthesized. The new compound series was characterized by 1H NMR and EIMS (Figures S1–S4 in Supporting Information File 1). In the DFT-optimized structures of 1, the ureylene moiety and the phenyl groups on both
- effects were previously reported for bis(NAr3) derivatives having π-conjugated bridges [8][9]. Judging from the long N···N distances (e.g., 13.16 Å for 1b+) in the DFT-optimized structure (Figure S5 in Supporting Information File 1), the through-space electrostatic interactions between the NAr3 units
- for 1b. The larger splitting for 1b with respect to 1a is a common feature between DFT calculations and electrochemical investigations. This indicates that electronic interactions between the NAr3 units largely contribute to the experimentally proven thermodynamic stability of the MV state. Indeed






Aggregation-induced emission effect on turn-off fluorescent switching of a photochromic diarylethene
Beilstein J. Org. Chem. 2019, 15, 2204–2212, doi:10.3762/bjoc.15.217

- functional theory (DFT) and time-dependent DFT (TDDFT). The excitation wavelengths as well as the emission wavelength qualitatively agrees with the experimental results. Since compound 1 consists of a diarylethene moiety and an imidazo[1,2-a]pyridine moiety, the characteristic of 1 has the combination of
- nm) and AS ONE Handy UV Lamp SLUV-4 (λ = 365 nm) were used. For visible light irradiation, a 500W USHIO SX-UI501XQ Xenon lamp attached with Toshiba color filters (Y-48, Y-44, and UV-29) was used. The Gaussian09 program package [32] was used for geometry optimizations with DFT for ground states and
- subsequent TDDFT calculations. For the calculation of the fluorescence, the geometry optimizations were performed for the first excited state obtained by TDDFT. The hybrid B3LYP functional [33][34][35] was adopted to exchange-correlation term of DFT. The gaussian 6-31G(d,p) basis set was adopted to all








Azologization and repurposing of a hetero-stilbene-based kinase inhibitor: towards the design of photoswitchable sirtuin inhibitors
Beilstein J. Org. Chem. 2019, 15, 2170–2183, doi:10.3762/bjoc.15.214

- functional theory (DFT) to optimize the ground state equilibrium structures of (E)-2b, (Z)-2b, 8a and 8b, and used time-dependent DFT (TDDFT) and high-level correlated methods to obtain UV–vis absorption energies and oscillator strengths. To obtain the simulated absorption spectrum and λmax values
- temperature. Computational details: All calculations were carried out using the TURBOMOLE version 7.2 quantum chemistry package [67]. Geometry optimizations of all compounds in different conformers were carried out using DFT with PBE approximation to the exchange-correlation (XC) functional and employing the










Click chemistry towards thermally reversible photochromic 4,5-bisthiazolyl-1,2,3-triazoles
Beilstein J. Org. Chem. 2019, 15, 2161–2169, doi:10.3762/bjoc.15.213

- Supporting Information File 1). The absorption spectral properties are summarized in Table 1 together with their predicted absorption maxima obtained by TD DFT calculations in vacuum [35]. Although 1o and 2o showed substantial coloration, 3o showed only a slight coloration at room temperature. The solvent
- observation was well-reproduced by TD DFT calculations (Table 1 and chapter SI-4 in Supporting Information File 1). When the absorption maximum wavelength of 1c, whose central ethene moiety is triazole, is compared with those of the closely related 7c (cyclopentene) [37], 8c (hexafluorocyclopentene) [38], 9c
- bond length nor its bond order, which were obtained by DFT calculations of these compounds, did not give a clear explanation for the difference in the reaction rate. Possible evidence of the fast back reaction of 3c may be found in the bond lengths and Mulliken bond orders between the atoms









Friedel–Crafts approach to the one-pot synthesis of methoxy-substituted thioxanthylium salts
Beilstein J. Org. Chem. 2019, 15, 2105–2112, doi:10.3762/bjoc.15.208

- examined using MeCN, CH3NO2, DMSO and MeOH, no substantial shifts of the main peak at around 460 nm in the UV–vis absorption spectra were observed [24], indicating that the main absorption of these catalysts would be due to π–π* transition, which is supported by DFT calculations (TD-DFT B3LYP method
- electron-withdrawing groups can be applied to the reaction. It was found that the main absorption of thioxanthylium salts around 460 nm in UV–vis spectra would be due to π–π* transitions, which was supported by DFT calculations. The present reaction provides a versatile access to functionalized
- equiv) and TfOH (2.0 equiv) were used at 120 °C. c2 (2.0 equiv) and TfOH (2.0 equiv) were used. d20 h. e2 h. The UV–vis spectra of thioxanthylium salt (0.1 mM) in CH3CN. Frontier orbitals of thioxanthylium salts, calculated by DFT at the B3LYP/6-31G(d,p) level of Orca. (a) LUMO localization of 3a








α-Photooxygenation of chiral aldehydes with singlet oxygen
Beilstein J. Org. Chem. 2019, 15, 2076–2084, doi:10.3762/bjoc.15.205

- dichroism (ECD) and TD-DFT methods. Keywords: 1,2-diols; ECD; enamines; organocatalysis; porphyrins; silyl ethers of diarylprolinols; singlet oxygen; Introduction Carbonyl compounds are one of the most important building blocks in organic synthesis. As a consequence, there is a constant need for new
- starting material and the catalyst. Results and Discussion Our previous studies on α-photooxygenation of achiral aldehydes with 1O2 in the presence of chiral amines supported by DFT calculations indicate that the reaction is highly enantioselective only when both enamine structural fragments (substituents
- anti’-6 obtained from the reaction with diarylprolinol silyl ether (R)-18 (Table 1, entry 5) were analyzed using electronic circular dichroism spectroscopy (ECD) and TD-DFT methods. So-called in situ methodology with dimolybdenum tetraacetate (19) acting as auxiliary chromophores which proved a very








1,2,3,4-Tetrahydro-1,4,5,8-tetraazaanthracene revisited: properties and structural evidence of aromaticity loss
Beilstein J. Org. Chem. 2019, 15, 2059–2068, doi:10.3762/bjoc.15.203

- led to variation of its electron donor/acceptor capability that allowed fine-tuning the absorption properties. The propensity of these compounds and a number of its derivatives to form infinite chains involving >N–H···N= and >N–H···Hal−···N+ atoms is demonstrated by X-ray structure analysis. The DFT
- fluorescent. Minimum two Pekarian functions are needed to reproduce the band shape of the salts 6 in ethanol indicating that at least two electronic transitions are involved (Supporting Information File 1, Figure S3). TD DFT calculations interprets the longest wavelength absorption bands of 3 and 6 to be
- predominantly the charge transfer type HOMO -> LUMO transitions (Supporting Information File 1, Tables S1 and S2). The easiness of protonation and stability of salts 6 evidenced the behavior of THTAA (3) as a strong base. Indeed, our DFT calculations of the proton affinity of THTAA (3) produced ΔH298 = 235.9















Bipolenins K–N: New sesquiterpenoids from the fungal plant pathogen Bipolaris sorokiniana
Beilstein J. Org. Chem. 2019, 15, 2020–2028, doi:10.3762/bjoc.15.198

- for each molecule was further optimised by DFT at the B3LYP-D3/def2-TZVPP level of theory using Turbomole 7.1 [38] and ECD spectra were calculated in Turbomole using TDDFT (B3LYP-D3/def2-TZVPP). Structures of compounds 1–12 isolated from B. sorokiniana. Key 2D NMR correlations of bipolenins K–N (1–4






Fluorinated azobenzenes as supramolecular halogen-bonding building blocks
Beilstein J. Org. Chem. 2019, 15, 2013–2019, doi:10.3762/bjoc.15.197
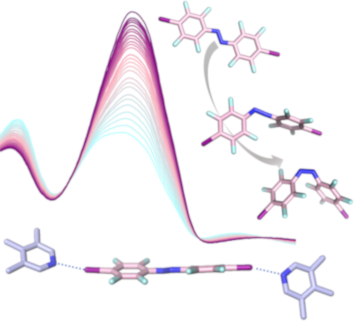
- azobenzenes with different halogen bonding donor properties are discussed in relation to their changing photophysical properties, rationalized by DFT calculations. Keywords: azobenzene; DFT calculations; fluorine chemistry; halogen bonding; photochemistry; Introduction The halogen bond is an attractive





A review of the total syntheses of triptolide
Beilstein J. Org. Chem. 2019, 15, 1984–1995, doi:10.3762/bjoc.15.194

- -endo-trig cyclization of 2-alkenyl-1,3-dithiolanes to access trans-decalins (Figure 2, route K) [78]. Density functional theory calculation (DFT) studies indicated that the 2-alkenyl-1,3-dithiolane moiety acts as a latent initiator, which triggers the cationic 6-endo-trig cyclization in the presence of











Reactions of 2-carbonyl- and 2-hydroxy(or methoxy)alkyl-substituted benzimidazoles with arenes in the superacid CF3SO3H. NMR and DFT studies of dicationic electrophilic species
Beilstein J. Org. Chem. 2019, 15, 1962–1973, doi:10.3762/bjoc.15.191

- -arylmethyl-substituted benzimidazoles, in yields up to 90%. The reaction intermediates, protonated species derived from starting benzimidazoles in TfOH, were thoroughly studied by means of NMR and DFT calculations and plausible reaction mechanisms are discussed. Keywords: benzimidazoles; cations; Friedel
- the formation of heteroaromatic benzyl-type dications IV, VI, and IX, respectively. Electronic characteristics, energies of HOMO/LUMO, electrophilicity indices ω [25][26], charge distribution, and contribution of atomic orbital into the LUMO of species I–IX were calculated by DFT method to estimate
- water. According to DFT calculations, protonation of the benzimidazole nitrogen N3 and the oxygen of the carbonyl or hydroxymethyl group in 1–8 leading to dicationic species I–III, V, VII, and VIII is thermodynamically favorable (−18.6 to −25.8 kcal/mol, Table 1). On the other hand, the dehydration of








Tautomerism as primary signaling mechanism in metal sensing: the case of amide group
Beilstein J. Org. Chem. 2019, 15, 1898–1906, doi:10.3762/bjoc.15.185

- ultrafine grid in the computation of two-electron integrals and their derivatives. The true minima were verified by performing frequency calculations in the corresponding environment. The TD-DFT method [27][28][29], carried out with the same functional and basis set, was used for predicting vertical









Inherent atomic mobility changes in carbocation intermediates during the sesterterpene cyclization cascade
Beilstein J. Org. Chem. 2019, 15, 1890–1897, doi:10.3762/bjoc.15.184

- these two methyl groups are critical for the preorganization of GFPP in the biosynthetic pathways leading to sesterfisherol and quiannulatene. Keywords: biosynthesis; carbocation; DFT; substrate recognition; terpene cyclase; Introduction Terpene synthases are thought to have four main roles: (i
- 5/6/8/5 tetracyclic intermediate. This, in turn, is transformed to a 4/6-membered ring in quiannulatene biosynthesis, whereas 5/5 ring formation proceeds in sesterfisherol biosynthesis (Scheme 1, Scheme 2, and Scheme 3). Based on our DFT calculations, this regioselectivity is determined by the







Halide metathesis in overdrive: mechanochemical synthesis of a heterometallic group 1 allyl complex
Beilstein J. Org. Chem. 2019, 15, 1856–1863, doi:10.3762/bjoc.15.181

- this and several related points more quantitatively, various features of the K/Cs/[allyl]− system were modeled with DFT calculations, using the B3PW91 hybrid functional [27][28] with Grimme’s -D3 dispersion corrections (GD3BJ) [29]. A calculation on the simple model systems [K(C3H5)] and [Cs(C3H5
- Information File 79: Experimental and computational details; crystal data and summary of X–ray data collection. Supporting Information File 80: Coordinates of DFT-optimized structures. Acknowledgements Financial support from the National Science Foundation (CHE-1665327) and the American Chemical Society




Complexation of 2,6-helic[6]arene and its derivatives with 1,1′-dimethyl-4,4′-bipyridinium salts and protonated 4,4'-bipyridinium salts: an acid–base controllable complexation
Beilstein J. Org. Chem. 2019, 15, 1795–1804, doi:10.3762/bjoc.15.173

- derivatives for the additional multiple hydrogen bonding interactions between the hosts and the guests, which were evidenced by 1H NMR titrations, X-ray crystal structures and DFT calculations. Moreover, it was also found that the association constants (Ka) of the complexes could be significantly enhanced
- by the additional multiple hydrogen-bonding interactions between the hosts and the guests, which were evidenced by 1H NMR titration, X-ray crystal structures and DFT calculations. Moreover, we also found that the Ka values of the complexes could be significantly enhanced with larger counteranions of
- main reason for the formation of the stable complex H5·G1. DFT calculation of host–guest complexes To further investigate the complexation mode and structural characteristics of the host–guest complexes, DFT calculations were carried out at the B3LYP/6-31G level of theory for complex H4·G1 (Supporting









Functional panchromatic BODIPY dyes with near-infrared absorption: design, synthesis, characterization and use in dye-sensitized solar cells
Beilstein J. Org. Chem. 2019, 15, 1758–1768, doi:10.3762/bjoc.15.169

- were computed by density functional theory modelling (DFT) and characterized through UV–vis spectroscopy and cyclic voltammetry (CV) measurements. Finally, we report preliminary results obtained using these functional dyes as photosensitizers in dye-sensitized solar cells (DSSCs). Keywords: boron
- application in DSSCs, in rather good agreement with the values obtained from DFT calculations. Finally, we report preliminary results employing these molecules as photosensitizers in dye solar cells with iodine-based liquid electrolytes. We show that the limited performances of these new BODIPY derivatives
- DFT calculations In many opto-electronic devices the light-absorption properties of the semiconductors are a critical parameter. This is particularly the case when solar energy conversion applications are targeted. For instance, in order to maximize the photocurrent density in a DSSC device, the








Synthesis, photophysical and electrochemical properties of pyridine, pyrazine and triazine-based (D–π–)2A fluorescent dyes
Beilstein J. Org. Chem. 2019, 15, 1712–1721, doi:10.3762/bjoc.15.167

- , Lippert–Mataga plots, cyclic voltammetry and density functional theory (DFT) calculations, we reveal the photophysical and electrochemical properties of the (D–π–)2A fluorescent dyes OUY-2, OUK-2 and OUJ-2. Results and Discussion Synthesis The (D–π–)2A fluorescent dyes OUY-2 [2], OUK-2 [3][4] and OUJ-2
- , OUK-2 and OUJ-2, respectively, estimated from DFT calculation at the B3LYP/6-31G(d,p) level of theory [47]), Δf is the orientation polarizability, ε is the static dielectric constant, and n is the refractive index of the solvent. The Lippert–Mataga plots (Figure 3) for the three dyes show high
- molecular structures and the molecular orbitals of the three dyes were calculated using the DFT at the B3LYP/6-31G(d,p) level of theory [47]. The results of the DFT calculation for the three dyes indicated that the HOMO is mostly localized on the two (diphenylamino)carbazole moieties containing the








Water inside β-cyclodextrin cavity: amount, stability and mechanism of binding
Beilstein J. Org. Chem. 2019, 15, 1592–1600, doi:10.3762/bjoc.15.163

- thermodynamic characteristics of β-CD hydration compare with those of its smaller α-cyclodextrin (α-CD) counterpart? In this study, we address these questions by employing a combination of experimental (DSC/TG) and theoretical (DFT) approaches. Keywords: β-cyclodextrin; DFT calculations; DSC/TG experiments
- smaller α-CD counterpart? In this study, we endeavor to address these questions by employing a combination of experimental (DSC/TG) and theoretical approaches (DFT). Our findings illuminate the mechanism of β-CD hydration at an atomic level and disclose the major factors controlling this process. A
- experimental) approaches are sometimes conflicting and contradictory. There is no “standard” method to investigate the complexation behavior of CDs. Thus, in our study a computational quantum mechanical modelling method, namely density functional theory (DFT) was chosen to investigate the electronic structure







Transient and intermediate carbocations in ruthenium tetroxide oxidation of saturated rings
Beilstein J. Org. Chem. 2019, 15, 1552–1562, doi:10.3762/bjoc.15.158

- tetroxide-mediated oxidation of cyclopentane, tetrahydrofuran, tetrahydrothiophene and N-substituted pyrrolidines has been studied computationally by DFT and topological (analysis of the electron localization function, ELF) methods. In agreement with experimental observations and previous DFT calculations
- carbocation. In the case of pyrrolidines, the carbocation is completely stabilized as an energy minimum in the form of an iminium ion and the reaction takes place in two steps. Keywords: alkanes; carbocations; DFT; oxidations; ruthenium tetroxide; Introduction Ruthenium-catalyzed oxidations [1][2] and, in
- Waegell et al. proposed a new (3 + 2) asynchronous concerted mechanism [19][20], both groups converged to the latter proposed mechanism when Bakke et al. changed the interpretation of their kinetic isotopic experiments [21][22][23][24]. The (3 + 2) concerted mechanism was further confirmed by DFT












An azobenzene container showing a definite folding – synthesis and structural investigation
Beilstein J. Org. Chem. 2019, 15, 1534–1544, doi:10.3762/bjoc.15.156

- unit as well as commercially available compounds. Investigation of the structure and the switching process To investigate the structures of the foldable container 10 in the gas phase, the geometric parameters of the trans,trans-, cis,trans- and cis,cis-isomers were fully optimized by means of the DFT
- Cotton effect in this region [43][49]. If this is taken into account, it becomes obvious that the cis,cis-isomer adopts the M,M conformation. This is in line with the DFT calculations finding only the cis,cis-(M,M) isomer as minimum on the energy potential surface. The spectrum of cis,trans-10 allows the
- performed by using the program package Gaussian 16 [62]. The geometries of the molecules were fully optimized in the gas phase by using the DFT potentials B3LYP [53][54][55] and B3LYP-D3 [56][57] as well as the 6-31G* [58][59] basis set. For all calculations, the default thresholds implemented in Gaussian










Superelectrophilic carbocations: preparation and reactions of a substrate with six ionizable groups
Beilstein J. Org. Chem. 2019, 15, 1515–1520, doi:10.3762/bjoc.15.153

- neighboring positions, while in the case of 16, the five cationic charges are separated into groups of three and two charges. The increased stability of the separated cationic charge is evident in the DFT calculated energies of the ions. At the B3LYP 6-311G (d,p) level, ion 16 is calculated to be 32.7
- from diol 9. Proposed mechanisms leading to products 10 and 11. Products and relative yields from the reaction of alcohol 18 with CF3SO3H and C6H6 [12]. Comparison of superelectrophilic carbocations (3–5 and 14) and their chemistry. DFT calculated relative energies of pentacations 16 and 21 [14








Different reactivity of phosphorylallenes under the action of Brønsted or Lewis acids: a crucial role of involvement of the P=O group in intra- or intermolecular interactions at the formation of cationic intermediates
Beilstein J. Org. Chem. 2019, 15, 1491–1504, doi:10.3762/bjoc.15.151

- phosphonic acids and other phosphorus-containing compounds. Contrary to Brønsted acids, 3-methylbuta-1,2-dien-1-ylphosphonic dichloride [Cl2(O=)P–HC=C=CMe2] reacted with the Lewis acid AlCl3 in an intermolecular way forming noncyclic intermediates, which were investigated by NMR spectroscopy and DFT
- intermolecular hydroarylation of allenes bearing electron-withdrawing substituents. Plausible reaction mechanisms have been proposed on the basis of the investigated reactions, and NMR analysis and DFT studies of the intermediate cationic species. Keywords: aluminum chloride; cation; intermediate
- electrophilic activation with Brønsted or Lewis (super)acids, including reactions with arenes as π-nucleophiles, and investigation of intermediate cationic species by means of NMR and DFT calculations. Allenes used in this study are presented in Figure 1. We explored allenes having different substituents at the














