Search results
Search for "radical" in Full Text gives 752 result(s) in Beilstein Journal of Organic Chemistry. Showing first 200.
Recent advancements in iodide/phosphine-mediated photoredox radical reactions
Beilstein J. Org. Chem. 2023, 19, 1785–1803, doi:10.3762/bjoc.19.131

- review, we primarily focus on summarizing the recent advancements in inexpensive and readily available iodide/phosphine-mediated photoredox radical transformations. Keywords: annulation; decarboxylative; iodide/phosphine; photocatalytic; radical reaction; Introduction Over the past few decades
- , numerous remarkable breakthroughs and notable progresses have been achieved in the realm of photoredox catalysis [1][2][3]. This domain has profoundly transformed modern organic synthesis, resulting in a considerable surge in research efforts centered on free radical reactions [4]. Presently, photoredox
- provided a more sustainable and economically viable approach but also demonstrated excellent performance in various transformations. It had been successfully applied to a series of radical reactions, including trifluoromethylation, deaminative alkylation, and asymmetric versions of Minisci reactions


























Selectivity control towards CO versus H2 for photo-driven CO2 reduction with a novel Co(II) catalyst
Beilstein J. Org. Chem. 2023, 19, 1766–1775, doi:10.3762/bjoc.19.129

- that the energy barriers of the reactions shown in Equation 2 and Equation 3 are lowered. In fact, the formation of the radical anion CO2−· takes place at −1.9 V versus normal hydrogen electrode (NHE), while the proton-assisted reductions of CO2 to CO and formic acid happen at −0.53 V and −0.61 V
- (helping in the deprotonation of the radical cation BIH•+ formed after the reductive quenching of the PS), but also can actively assist the catalysis, by capturing CO2 [50][51][52]. On the other hand, having three hydroxy groups, TEOA is also considered a proton donor and the formation of metal hydrides is
- accumulation of the reduced PS− species. We propose the following mechanism (Scheme 1). The PS absorbs a photon (420 nm) and in its excited state is quenched by BIH, which is deprotonated by the base (TEA) and forms a radical (BI·). Since this radical is highly reducing, it can happen that this species can







Benzoimidazolium-derived dimeric and hydride n-dopants for organic electron-transport materials: impact of substitution on structures, electrochemistry, and reactivity
Beilstein J. Org. Chem. 2023, 19, 1651–1663, doi:10.3762/bjoc.19.121

- strength and their reactivity with organic semiconductors (SC) does not depend solely on the SC reduction potential, since the first step, at least in many cases, is a hydride transfer rather than an electron transfer [8][9]. Moreover, as well forming the desired semiconductor radical anion SC•−, and the
- presumably dominant in the present case. The importance of radical stabilization may in part be because the positive charges in Y = H or alkyl 1+ ions is already substantially stabilized by the aromaticity of the benzimidazolium ions, whereas the spin densities of the corresponding 1• radicals are highly
- ring [57]. Presumably inductive effects destabilizing 1i+, different extents of planarization, and improved radical stabilization by the 5-(dimethylamino)-2-thienyl susbtituent play a role. As expected, R' = OMe groups on the six-membered benzimidazolium ring do have a net cation-stabilizing effect










Tying a knot between crown ethers and porphyrins
Beilstein J. Org. Chem. 2023, 19, 1630–1650, doi:10.3762/bjoc.19.120

- 1.70–2.50 ns. An apparent colour change was observed upon treatment of 42 with AgSbF6 and CuCl2, indicating radical cation formation 42•+. ESR spectra and coulometric oxidation experiments further supported the presence and stability of the radical species. The reactions of 38 with a pre-functionalized




























Radical chemistry in polymer science: an overview and recent advances
Beilstein J. Org. Chem. 2023, 19, 1580–1603, doi:10.3762/bjoc.19.116

- Zixiao Wang Feichen Cui Yang Sui Jiajun Yan School of Physical Science and Technology, ShanghaiTech University, 393 Middle Huaxia Rd., Shanghai, 201210, China 10.3762/bjoc.19.116 Abstract Radical chemistry is one of the most important methods used in modern polymer science and industry. Over the
- past century, new knowledge on radical chemistry has both promoted and been generated from the emergence of polymer synthesis and modification techniques. In this review, we discuss radical chemistry in polymer science from four interconnected aspects. We begin with radical polymerization, the most
- employed technique for industrial production of polymeric materials, and other polymer synthesis involving a radical process. Post-polymerization modification, including polymer crosslinking and polymer surface modification, is the key process that introduces functionality and practicality to polymeric






















C–H bond functionalization: recent discoveries and future directions
Beilstein J. Org. Chem. 2023, 19, 1568–1569, doi:10.3762/bjoc.19.114

- well as famous Noble-prize-winning cross-couplings, therefore approaching another step up towards sustainability. Likewise, a free-radical process is also a classical way to functionalize nonactivated C−H bonds in which site selectivity arises either from the relative strength of the C−H bonds or from
- the abstraction of intramolecular hydrogen atoms. Radical chemistry is a viable alternative to the two-electron process, involving C–H bond functionalization in the absence of any ligand and using low-cost redox-active metals (Fe, Cu, Mn, etc.) rather than heavy metals (Rh, Ir, etc.). Although radical
Lewis acid-promoted direct synthesis of isoxazole derivatives
Beilstein J. Org. Chem. 2023, 19, 1562–1567, doi:10.3762/bjoc.19.113

- that the reaction is not a free radical reaction. Based on the control experiments and previous literature [21], we propose the following possible mechanism, which is shown in Scheme 5. Aluminum trichloride reacts with sodium nitrite to form an intermediate aluminum complex A, which is further








Synthesis of 5-arylidenerhodanines in L-proline-based deep eutectic solvent
Beilstein J. Org. Chem. 2023, 19, 1537–1544, doi:10.3762/bjoc.19.110

- screenings and they present a wide spectrum of pharmacological activities [1][2]. Thus, for example, 3,4-dihydroxybenzylidenerhodanine (A) showed a high antioxidant activity with 71.2% of 1,1-diphenyl-2-picrylhydrazyl radical (DPPH) scavenging activity [3]. Naphthalen-2-ylmethylidenerhodanine (B) has been
- recycled for several runs [20]. As some benzylidenerhodanine derivatives were already reported for their antioxidant activities [3], we investigated those compounds for their antioxidant activity expressed as percentage of 1,1-diphenyl-2-picrylhydrazyl radical (DPPH) scavenging activity. DPPH free radicals
- hydroxy group showed DPPH radical scavenging activity. Compound 3d with a catechol-like structure exhibited the best antioxidant activity. Experimental General procedure for the Knoevenagel condensation DES (0.8 g) was introduced in a 10 mL round-bottomed flask. Then, the aldehyde (0.5 mmol) and rhodanine



N-Sulfenylsuccinimide/phthalimide: an alternative sulfenylating reagent in organic transformations
Beilstein J. Org. Chem. 2023, 19, 1471–1502, doi:10.3762/bjoc.19.106

- ·H2O, FeSO4, and Fe(acac)3 resulted in inferior chemical yields. Employment of 2,2,6,6-tetramethylpiperidinyl-1-oxyl (TEMPO) as a radical trapper inhibited the reaction, which proved that a radical process was involved. The reaction was initiated by a single electron transfer (SET) process from the
- sulfur atom to Fe3+ to generate Fe2+ and radical cation I. Subsequent cleavage of the N–S bond led to cation II and radical III. Interaction of III with Fe2+ regenerated the Fe3+ species and IV. At the same time, electrophilic addition of II to alkene 9 yielded intermediate V, which was subjected to the
- Scheme 31. Initially, homolytic cleavage of thiosulfonate 70 generated PhS· and PhSO2· radicals. The reduction of Ni(II) to Ni(0) in the presence of Cs2CO3 and the reaction with 68 formed alkynyl-Ni species I. Then, the PhS· radical reacted with I to generate alkenyl radical II, which can react with the










































































α-(Aminomethyl)acrylates as acceptors in radical–polar crossover 1,4-additions of dialkylzincs: insights into enolate formation and trapping
Beilstein J. Org. Chem. 2023, 19, 1443–1451, doi:10.3762/bjoc.19.103

- that α-(aminomethyl)acrylates are suitable acceptors for 1,4-additions of dialkylzincs in aerobic conditions. The air-promoted radical–polar crossover process involves the 1,4-addition of an alkyl radical followed by homolytic substitution at the zinc atom of dialkylzinc. Coordination of the nitrogen
- atom to zinc enables this SH2 process which represents a rare example of alkylzinc-group transfer to a tertiary α-carbonyl radical. The zinc enolate thus formed readily undergoes β-fragmentation unless it is trapped by electrophiles in situ. Enolates of substrates having free N–H bonds undergo
- levels of chiral induction, paving the way to enantioenriched β2-amino acids and β2,2-amino acids. Keywords: β-amino acids; tandem reactions; radical–polar crossover; tert-butanesulfinamide; zinc radical transfer; Introduction Dialkylzinc reagents react in aerobic medium with a range of α,β-unsaturated










Application of N-heterocyclic carbene–Cu(I) complexes as catalysts in organic synthesis: a review
Beilstein J. Org. Chem. 2023, 19, 1408–1442, doi:10.3762/bjoc.19.102

- primary, secondary, and tertiary alkyl halides. The mechanistic investigation revealed the generation of a silyl–copper intermediate which activates the alkyl halides by a single electron transfer to form alkyl radical intermediates [54]. It was suggested that substituting B2pin2 for PhMe2Si-Bpin would






















































































Visible-light-induced nickel-catalyzed α-hydroxytrifluoroethylation of alkyl carboxylic acids: Access to trifluoromethyl alkyl acyloins
Beilstein J. Org. Chem. 2023, 19, 1372–1378, doi:10.3762/bjoc.19.98

- ., Zibo 256401, China 10.3762/bjoc.19.98 Abstract A visible-light-induced nickel-catalyzed cross coupling of alkyl carboxylic acids with N-trifluoroethoxyphthalimide is described. Under purple light irradiation, an α-hydroxytrifluoroethyl radical generated from a photoactive electron donor–acceptor
- nickel-catalyzed coupling of aryl bromides with an α-hydroxytrifluoroethyl radical for the synthesis of trifluoromethyl aryl alcohols [39]. Encouraged by this work, we envisioned that the nickel-catalyzed coupling of carboxylic acids-derived acyl electrophiles with an α-hydroxytrifluoroethyl radical
- light-induced charge transfer event to give trifluoroethoxyl radical B, followed by a 1,2-hydrogen atom transfer (HAT), producing the stable radical C. For the nickel cycle, it is initiated by oxidative addition of Ni(0) catalyst E to acyl electrophile D formed in situ from carboxylic acid 1 with





Synthesis of ether lipids: natural compounds and analogues
Beilstein J. Org. Chem. 2023, 19, 1299–1369, doi:10.3762/bjoc.19.96































































































Non-noble metal-catalyzed cross-dehydrogenation coupling (CDC) involving ether α-C(sp3)–H to construct C–C bonds
Beilstein J. Org. Chem. 2023, 19, 1259–1288, doi:10.3762/bjoc.19.94

- oxidant to generate an oxygen-radical cationic intermediate, which undergoes abstraction of a hydrogen radical (or loses a proton first, followed by an electron) to afford an oxonium ion intermediate. Finally, the oxonium ion is attacked by various nucleophiles to obtain the target functionalized product
- . Route b: the α-C(sp3)–H bonds are activated by a combination of transition metals and radical initiators to give the alkyl radicals, which are coupled with other radical receptors to afford the target product. Cu-catalyzed reactions Copper (common oxidation states are +I, +II and +III) has a
- , tetrahydrofuran or tetrahydropyran can smoothly react with many methyl aryl ketones to obtain the desired coupling products (Scheme 6a) [54]. The mechanism of the dehydrogenation cross-coupling reaction may undergo a radical pathway. Initially, the tert-butoxy radical produced by the dissociation of t-BuOOH may












































Radical ligand transfer: a general strategy for radical functionalization
Beilstein J. Org. Chem. 2023, 19, 1225–1233, doi:10.3762/bjoc.19.90

- challenging-to-generate “uncontrollable” species prone to side reactions to versatile reactive intermediates enabling construction of myriad C–C and C–X bonds. This maturation of free radical chemistry has been enabled by several advances, including the proliferation of efficient radical generation methods
- , such as hydrogen atom transfer (HAT), alkene addition, and decarboxylation. At least as important has been innovation in radical functionalization methods, including radical–polar crossover (RPC), enabling these intermediates to be engaged in productive and efficient bond-forming steps. However, direct
- engagement of alkyl radicals remains challenging. Among these functionalization approaches, a bio-inspired mechanistic paradigm known as radical ligand transfer (RLT) has emerged as a particularly promising and versatile means of forming new bonds catalytically to alkyl radicals. This development has been






Enabling artificial photosynthesis systems with molecular recycling: A review of photo- and electrochemical methods for regenerating organic sacrificial electron donors
Beilstein J. Org. Chem. 2023, 19, 1198–1215, doi:10.3762/bjoc.19.88
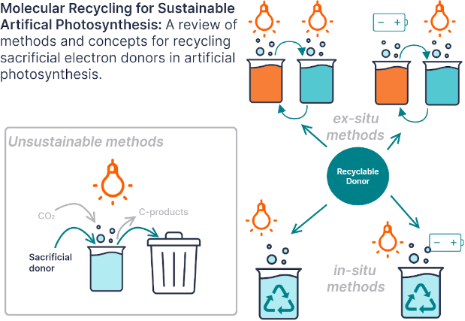
- Figure 4 can be reduced to a stable radical and reoxidized in aqueous media [65]. Depending on the pH, the reduction potential to form the 2 electron-reduced species is close to that of the stable radical. In non-aqueous media analogues of this species form stable 2 electron-reduction products. In
- candidates for electron donors in systems that require PCET because PCET prevents dimerizable radical formation. However, a too low pH value could be detrimental. The redox potentials recorded for BNAH were also reported to be very sensitive to the electrode material. Many carbon dioxide reduction systems
- -Dimethylamino)toluene (DMT), has been used as a sacrificial electron donor in artificial photosynthesis [3]. The radical species that forms after oxidation can dimerize by forming a carbon–carbon bond which cannot be broken by re-reduction [3][73]. Voltammetric studies to identify the byproducts of DMT






Exploring the role of halogen bonding in iodonium ylides: insights into unexpected reactivity and reaction control
Beilstein J. Org. Chem. 2023, 19, 1171–1190, doi:10.3762/bjoc.19.86

- -effect; Introduction Iodonium ylides are a subset of hypervalent iodine (HVI) reagents that were first reported in 1957 by Neiland [1]. These have since been investigated under a variety of thermal, photochemical, radical and transition metal-catalyzed conditions [2], and they have been successfully
- intermediate was not viable under such mild conditions. The initially proposed ionic pathway (Figure 5, left) was abandoned as solvent effects had little influence on the reaction rate, and since no Wagner–Meerwein rearrangement products were detected with bicyclic olefin precursors. Radical-based pathways
- proposed that electron donor–acceptor (EDA) complex 36 was initially formed between 32 and a sacrificial equivalent of 31, and that 36 underwent a SET to give radical anion 37 and radical cation 38 (Figure 8). While one equivalent of the ylide orchestrated a series of proton transfer (PT) and SET events



































Selective and scalable oxygenation of heteroatoms using the elements of nature: air, water, and light
Beilstein J. Org. Chem. 2023, 19, 1146–1154, doi:10.3762/bjoc.19.82

- oxygen- or radical-sensitive functionalities (i.e., an amino (2w) or nitro group (2x)). On the other hand, oxidizable groups, such as alcohols (2e), and halogens, such as such as chloro and fluoro on the aromatic ring (2i ,2j, 2k), were well tolerated. However, the presence of a iodo group (2v
- originates from water. In this tentative mechanism, the sulfide I forms with water and oxygen a photoactive complex II which is excited at 365 nm towards III. Via single-electron transfer both a radical cation IV and the superoxide V are generated. Subsequently, the sulfide radical cation IV undergoes a
- nucleophilic attack by water. The superoxide first abstracts a proton to form the perhydroxyl radical VII followed by hydrogen atom abstraction from intermediate VIII to yield sulfoxide IX. The generated hydrogen peroxide decomposes into water and oxygen. The novel proposed pathway can either be dominant or







Photoredox catalysis harvesting multiple photon or electrochemical energies
Beilstein J. Org. Chem. 2023, 19, 1055–1145, doi:10.3762/bjoc.19.81

- the most appropriate for a given reaction, scale and purpose of a project. Keywords: consecutive photoinduced electron transfer; electro-activated photoredox catalysis; photoelectrochemistry; photoredox catalysis; radical ions; Review 1 Introduction Owing to the unique reactivity patterns of free
- intermediate is proposed in conPET and PEC reactions (e.g., a photoexcited radical anion), yet different reactivity outcomes arise; the underlying reasons for such are discussed. Finally, we provide our perspective on current challenges and target areas for future exploration. 1.1 Multi-photon processes As
- radical anion or radical cation. As a semi-stable, higher energy ground-state entity, this can accumulate in sufficient concentration under the reaction conditions to absorb another photon and thereby generate a super-reducing or super-oxidizing excited state (Figure 2 left). In addition to ‘radical ion













































































The effect of dark states on the intersystem crossing and thermally activated delayed fluorescence of naphthalimide-phenothiazine dyads
Beilstein J. Org. Chem. 2023, 19, 1028–1046, doi:10.3762/bjoc.19.79

- nm) [39], while this positive absorption band is closer to that of NI−•, which was determined at ca. 430 nm [58]. Therefore, this absorption band is assigned to the NI radical anion. The excited state absorption (ESA) band centered at 460 nm is attributed to the 3NI state. Moreover, another positive















Photoredox catalysis enabling decarboxylative radical cyclization of γ,γ-dimethylallyltryptophan (DMAT) derivatives: formal synthesis of 6,7-secoagroclavine
Beilstein J. Org. Chem. 2023, 19, 918–927, doi:10.3762/bjoc.19.70

- Alessio Regni Francesca Bartoccini Giovanni Piersanti Department of Biomolecular Sciences, University of Urbino, Carlo Bo Piazza Rinascimento 6, 61029 Urbino, PU, Italy 10.3762/bjoc.19.70 Abstract An unusual photoredox-catalyzed radical decarboxylative cyclization cascade reaction of γ,γ
- easily generate reactive open-shell radical species and/or intermediates. The substrate is consequently activated for bond cleavage, atom abstraction, or nucleophilic or electrophilic attack. After quenching, the oxidized or reduced photocatalyst regains or loses an electron to return to the starting
- selectively targeted by photoredox catalysis to enable unprecedented modification of the amino acid. In this context, it is worth mentioning that the single-electron oxidation of the indole moiety in tryptophan provides the radical cation, which enables selective C-radical generation at the weaker benzylic








Synthesis of aliphatic nitriles from cyclobutanone oxime mediated by sulfuryl fluoride (SO2F2)
Beilstein J. Org. Chem. 2023, 19, 901–908, doi:10.3762/bjoc.19.68

- ][26][27][28][29], a synthesis method for δ-olefin-containing aliphatic nitriles by the radical C–C bond cleavage of cycloketone oxime ester derivatives was developed by Shi’s group (Scheme 2a) [30], which emerged as an efficient strategy to construct C(sp2)–C(sp3) bonds [31][32][33]. Later, Xiao [34
- successfully obtained in 45% yield, which indirectly proved the existence of an oxime sulfonyl ester intermediate (fluorosulfonate). As shown in Scheme 5b, in the presence of one equivalent of TEMPO, a commonly used radical scavenger, the yield of 3aa significantly decreased, in addition, the reaction was
- afford the iminyl radical intermediate II. In the following step, the ring-strain of cyclobutanone is released under the promotion of the imine radical, giving the C-centered radical III which is subsequently captured by the alkene. Meanwhile, the radical IV transfers an electron to [Cun+1] regenerating







Pyridine C(sp2)–H bond functionalization under transition-metal and rare earth metal catalysis
Beilstein J. Org. Chem. 2023, 19, 820–863, doi:10.3762/bjoc.19.62

- starting from both cyclic and acyclic alkyl bromides. The findings of the reaction’s stereochemistry and observations made during some cyclization or ring-opening reactions indicated that the C–H alkylation may proceed through a radical-type mechanism. Next, in 2013, Wang and co-workers [52] reported a
- N-oxide 119b was formed during benzylation of 2-ethylpyridine N-oxide. A possible mechanism has also been reported (Scheme 23b). Electrophilic palladation at the C2-position of pyridine N-oxide 9 provides intermediate 120. The radical intermediate 121 is generated in situ by H-atom abstraction from
- toluene 117 by sulfate radical anion. Coordination of intermediate 120 and 121 leads to complex 122 which undergoes reductive elimination to provide product 119. 2-Ethyl-substituted pyridine N-oxides may undergo a dual C–H activation due to the buttressing effect of the ethyl group to produce azafluorene


















































Sulfate radical anion-induced benzylic oxidation of N-(arylsulfonyl)benzylamines to N-arylsulfonylimines
Beilstein J. Org. Chem. 2023, 19, 771–777, doi:10.3762/bjoc.19.57

- -aryl(benzyl)amines to N-arylimines using K2S2O8 is reported to be problematic, the oxidation of N-(arylsulfonyl)benzylamines to N-arylsulfonylimines using K2S2O8 has been achieved for the first time. The dual role of the sulfate radical anion (SO4·−), including hydrogen atom abstraction (HAT) and
- single electron transfer (SET), is proposed to be involved in the plausible reaction mechanism. Keywords: arylsulfonylimine; benzylic oxidation; benzyl sulfonamide; K2S2O8; sulfate radical anion; Introduction Among various imine compounds [1], N-arylsulfonylimines are perhaps the most prominent due to
- abstraction (HAT) followed by single electron transfer (SET) enabled by the sulfate radical anion (SO4·−). Results and Discussion Initially, we investigated the reaction of N-benzenesulfonyl(benzyl)amine (1a) as a model substrate with K2S2O8 in MeCN at 80 °C for 12 h, conditions that were used earlier in our







Strategies in the synthesis of dibenzo[b,f]heteropines
Beilstein J. Org. Chem. 2023, 19, 700–718, doi:10.3762/bjoc.19.51

- the enol carboxylate and subsequent 1,2 radical rearrangement and decarboxylation. Moderate to good yields of dibenzo[b,f]oxepine carboxylates 25 were achieved (63–85%). Stopka et al. [46] reported on tandem C–H functionalisation and ring expansion as an alternative to the Wagner–Meerwin rearrangement










































