Search results
Search for "calculations" in Full Text gives 763 result(s) in Beilstein Journal of Organic Chemistry. Showing first 200.
Ring-closing metathesis of prochiral oxaenediynes to racemic 4-alkenyl-2-alkynyl-3,6-dihydro-2H-pyrans
Beilstein J. Org. Chem. 2020, 16, 2757–2768, doi:10.3762/bjoc.16.226

- , 372.1570; found, 372.1571. Computational details DFT calculations were performed using the Gaussian 16 program suite [43]. For visualizations, the GaussView 6 interface program was used [44]. Due to economy, the pure M06L functional [45], def2-SV(P) basis set [46], and resolution of identity approximation























Synthesis of purines and adenines containing the hexafluoroisopropyl group
Beilstein J. Org. Chem. 2020, 16, 2739–2748, doi:10.3762/bjoc.16.224

- that with the CF3 moieties nearer the five-membered ring of the azabenzimidazole unit, and the rotamer of higher energy was that with the CF3 moieties near the six-membered ring (see Figure 4). Density functional theory calculations predict an enthalpy difference of 0.48 kcal⋅mol−1 between the two











Vicinal difluorination as a C=C surrogate: an analog of piperine with enhanced solubility, photostability, and acetylcholinesterase inhibitory activity
Beilstein J. Org. Chem. 2020, 16, 2663–2670, doi:10.3762/bjoc.16.216

- of 2 using the GIAO method with the B3LYP/6-311+G(d,p) level of theory. Chloroform was used as the solvent for both the NMR experiments and the SMD calculations. The three lowest-energy structures to emerge from the computational analysis (i.e., 2a–c) are shown in Figure 2. The global minimum
- approximate rotamers I and IV, respectively (Figure 1). The unexpectedly high relative energy that was calculated for compound 2f might be partially attributable to the fact that it features only a single F···H contact within 2.50 Å. The calculations suggest that the conformer 2f is not significantly








Water-soluble host–guest complexes between fullerenes and a sugar-functionalized tribenzotriquinacene assembling to microspheres
Beilstein J. Org. Chem. 2020, 16, 2551–2561, doi:10.3762/bjoc.16.207

- Ka = (2.20 ± 0.16) × 105 M−1, respectively. The binding affinity between TBTQ-(OG)6 and C60 was further verified by Raman spectroscopy. The geometry of the complex of TBTQ-(OG)6 with C60 deduced from DFT calculations indicates that the driving force of the complexation is mainly due to the
- diffraction. Therefore, the optimized geometry of the 1:1 complex of TBTQ-(OG)6 C60 in water was simulated by density functional theory (DFT) calculations at the B3LYP/6-31G(d) level of theory, which was completed with the aid of Molclus, MOPAC, and ORCA 4.1.0 programs [54][55][56]. As shown in Figure 6, C60
- were dried on a slide glass. C60 was tested in powder form on a slide glass. Molecular model of the complex TBTQ-(OG)6 C60 in water, as generated by DFT calculations. (a) Side-view; (b) top-view; (c) hydrophobic surface diagram. In part, H atoms were omitted for clarity (yellow: C, red: O, blue: N





![[Graphic 2]](/bjoc/content/inline/1860-5397-16-207-i3.svg?max-width=637&scale=1.18182) C60 [TBTQ-(OG)6: 50 μM; C60: 50 μM] and (b) TBTQ-(OG)6
C60 [TBTQ-(OG)6: 50 μM; C60: 50 μM] and (b) TBTQ-(OG)6 



Comparative ligand structural analytics illustrated on variably glycosylated MUC1 antigen–antibody binding
Beilstein J. Org. Chem. 2020, 16, 2540–2550, doi:10.3762/bjoc.16.206

- per the scripts provided with CHARMM-GUI, except for adjustments to the time step and number of iterations. The calculations were carried out using Nvidia V100 GPUs. The equilibration step included 5000 steps of minimization follows by 25000 steps of NVT dynamics (constant volume and temperature) with






Leveraging glycomics data in glycoprotein 3D structure validation with Privateer
Beilstein J. Org. Chem. 2020, 16, 2523–2533, doi:10.3762/bjoc.16.204

- – how many unique monosaccharides are modelled in the glycan chain, the total length of the glycan chain and computes the total number linkages between monosaccharides. After the composition calculations are carried out, the algorithm begins the generation of the notation by printing out the unit counts





NMR Spectroscopy of supramolecular chemistry on protein surfaces
Beilstein J. Org. Chem. 2020, 16, 2505–2522, doi:10.3762/bjoc.16.203

- though a total of 6 basic residues are present [80]. Most of the identified patches bound by the tweezers contain two basic residues. QM/MM calculations [7] show that one of these residues is bound inside the tweezer cavity while the second residue can form a salt bridge with the second phosphate group







Conformational preferences of fluorine-containing agrochemicals and their implications for lipophilicity prediction
Beilstein J. Org. Chem. 2020, 16, 2469–2476, doi:10.3762/bjoc.16.200

- conformers (II, VI, and VII), and with a rotatable C–C(F) bond that generates different conformers (I). The μ values for all herbicides were computed through theoretical calculations (see computational details section) and are presented in Table 2 along with their respective experimental log P data
- been successfully applied to predict the conformational energies of other fluorine-containing compounds [32][33][34]. Frequency calculations were performed to confirm that the optimized geometries were true energy minima (no imaginary frequency) and to estimate thermodynamic energies, at 298.15 K








Computational tools for drawing, building and displaying carbohydrates: a visual guide
Beilstein J. Org. Chem. 2020, 16, 2448–2468, doi:10.3762/bjoc.16.199

- computational tools was inspected for general features related to sketching, representing and model building, all of which could be further used as input for translation into other formats, search from glycan databases or complex calculations such as molecular simulations. Several tools feature an interactive
- models. These tools generally use 3D molecular templates of monosaccharides to reconstruct a 3D model. Energy minimisation methods can further refine the models. These models are essential for structure-based studies and complex calculations like Molecular Dynamics simulations. Therefore, the accurate












Recent developments in enantioselective photocatalysis
Beilstein J. Org. Chem. 2020, 16, 2363–2441, doi:10.3762/bjoc.16.197
















































































Synthesis of 1,4-benzothiazinones from acylpyruvic acids or furan-2,3-diones and o-aminothiophenol
Beilstein J. Org. Chem. 2020, 16, 2322–2331, doi:10.3762/bjoc.16.193

- Information File 376: Experimental details, copies of NMR spectra, X-ray crystallographic details, detailed antimicrobial and toxicity assays, results of semi-empirical calculations of partial charges. Acknowledgements The authors thank Danila Yu. Apushkin and Aleksandr I. Andreev (PSU, Perm, Russia) for










Styryl-based new organic chromophores bearing free amino and azomethine groups: synthesis, photophysical, NLO, and thermal properties
Beilstein J. Org. Chem. 2020, 16, 2282–2296, doi:10.3762/bjoc.16.189

- dyes bearing the julolidine donor as 1430 × 10−48 esu (for free amino derivative) and 1950 × 10−48 esu (for Schiff base derivative), respectively. The structural and electronic properties of the dyes as well as their NLO properties were further studied using DFT calculations. The thermal stabilities of
- all dyes were evaluated by thermogravimetric analysis (TGA). The TGA data showed that all dyes were thermally stable up to 250 °C. Keywords: DFT calculations; NLO; pH sensitive dyes; Schiff base; solvent effect; styryl dyes; Introduction Push-pull organic molecules are a class of organic dyes
- series of new styryl-based organic chromophores containing a free amino group and the corresponding Schiff base derivatives. The photophysical, pH sensitivity, NLO properties, and thermal stabilities of all synthesized dyes were investigated. Density Functional Theory (DFT) calculations were also












Synthetic approaches to bowl-shaped π-conjugated sumanene and its congeners
Beilstein J. Org. Chem. 2020, 16, 2212–2259, doi:10.3762/bjoc.16.186

- thermoelectric properties in addition to the onigiri-type core-shell assemblies have been reported for sumanene and its derivatives. More interestingly, its application in the absorption of small molecules such as NH3, CO2, CO, and H2 using density functional theory (DFT) calculations has also been revealed [25
- /mol) transformation of 12 (syn) to 17, calculated by density functional theory (DFT) calculations. On the other hand, in 2008, Higashibayashi et al. reported the synthesis of first chiral C3-symmetric trimethylsumanene 28 starting from enantiopure norbornadiene (10) by employing a rational synthetic
- (DFT) calculations. As we are aware that if the carbene formed is a singlet then a C–H bond inserted product is predominating whereas if the dimer is the major product along with the minor C–H bond inserted product then the triplet carbene is generated. During their study, they obtained the C–H






































































Photosensitized direct C–H fluorination and trifluoromethylation in organic synthesis
Beilstein J. Org. Chem. 2020, 16, 2151–2192, doi:10.3762/bjoc.16.183

- ) calculations [199]. They were able to experimentally demonstrate, for the first time, that the reaction between photoexcited AQN and Selectfluor® afforded a transient AQN–Selectfluor® triplet exciplex species by calculating the predicted TDDFT absorption spectrum that matched with the experimentally obtained
- TA spectra. The direct reaction between AQN and the substrate was not observed by TAS. The authors’ DFT calculations revealed reaction pathways that were thermodynamically and kinetically plausible. Initially, AQN and Selectfluor® (S0 in Scheme 16) form a van der Waals complex RC1, which is markedly






















































Lipophilicity trends upon fluorination of isopropyl, cyclopropyl and 3-oxetanyl groups
Beilstein J. Org. Chem. 2020, 16, 2141–2150, doi:10.3762/bjoc.16.182

- even for a set of relatively simple compounds, fragment-based clogP calculations are not guaranteed to give reliable lipophilicity data. Next, theoretical lipophilicities were obtained using DFT calculations, based on the notion that the partition coefficient of a given solute between two phases
- relates to the difference in Gibbs energy of the free ligand conformations of this solute in the respective solvents. Quantum chemistry calculations of Gibbs energies in octanol and water thus provide theoretical estimations of lipophilicities. Such estimations require systematic conformational analyses
- meaningless. This was a surprise, as series D and E are relatively rigid, which simplifies conformational analysis. The remarkably similar DFT-logP values suggest that the influence of the fluorination is underrepresented in the calculations. The following observations are noteworthy. The trifluorinated D5












GlypNirO: An automated workflow for quantitative N- and O-linked glycoproteomic data analysis
Beilstein J. Org. Chem. 2020, 16, 2127–2135, doi:10.3762/bjoc.16.180

- -specific or peptide-specific. If the user trusts Byonic’s site-specific assignment, then all peptide variants that contain that site are included in calculations of its occupancy and glycoform distribution. If the user prefers to perform a peptide-specific analysis, then each proteolytically unique peptide
- grouping: site-specific analysis, or peptide-specific analysis. For site-specific analysis, the site-specificity of glycosylation assigned by Byonic was trusted, and all peptide variants that contained that site were included in calculations of its occupancy and glycoform distribution. PSMs with identical






Muyocopronones A and B: azaphilones from the endophytic fungus Muyocopron laterale
Beilstein J. Org. Chem. 2020, 16, 2100–2107, doi:10.3762/bjoc.16.177

- experimental ECD spectra to elucidate the absolute configuration. In the quantum-chemical calculations, the generation of an excessive number of conformers was avoided using a molecular model in which the β-hydroxycarboxylic acid side chain at the C-7 position was simplified to an acetyloxy group (Figure 4A
- calculations were conducted at the B3LYP/6-311+G(d,p) level of theory. The calculated spectra were displayed using a Gaussian band shape with 0.28 eV. Antibacterial assays To evaluate the antibacterial activities of the isolated substances, five strains of Gram-positive bacteria were used, namely
- ) medium at a concentration of 1 × 106 CFU/mL. The recorded MIC was the lowest concentration at which no growth was observed. Structures of compounds 1–4. Key HMBC (green arrows) and COSY (bold) correlations in 1 and 2. Simplified model structures for calculations of the ECD spectra of 1. Comparison of the






Clustering and curation of electropherograms: an efficient method for analyzing large cohorts of capillary electrophoresis glycomic profiles for bioprocessing operations
Beilstein J. Org. Chem. 2020, 16, 2087–2099, doi:10.3762/bjoc.16.176

- for assessment. With large cohorts of CE-LIF data, peak picking and peak area calculations still remain a problem for fast and accurate quantitation, despite the presence of internal and external standards to reduce misalignment for the qualitative analysis. The peak picking and area calculation
- quantitation could be achieved using defined peak windows, migration time positions, and HappyTools peak area calculations (Figure 1C and 1D). Quantitation could be achieved with a simple user interface and HappyTools returned the glycan profile and quantitation results efficiently. Unfortunately, the
- automated quantitation with HappyTools (Gaussian mode) and other software were hampered by complications in peak picking and peak area calculations of non-Gaussian peaks (see next section). This required significant time (2–3 days) to manually inspect and correct the quantitative values of the peaks in the







Isolation and structure determination of a tetrameric sulfonyl dilithio methandiide in solution based on crystal structure analysis and 6Li/13C NMR spectroscopic data
Beilstein J. Org. Chem. 2020, 16, 2057–2063, doi:10.3762/bjoc.16.172

- negative hyperconjugation. Density functional theory and natural bond order calculations of dilithio methandiides of type II [32][43] suggest that the two lone pairs at the dianionic carbon atom of (2a)4·(THF)6 and (2a)6·Li2O·(THF)6 are perhaps located in an sp2 hybridized orbital and p orbital. However







How and why plants and human N-glycans are different: Insight from molecular dynamics into the “glycoblocks” architecture of complex carbohydrates
Beilstein J. Org. Chem. 2020, 16, 2046–2056, doi:10.3762/bjoc.16.171

- , namely the three gg, gt and tg conformations for each 1-6 torsion. The topology file for each structure was obtained using tleap [31], with parameters from the GLYCAM06-j1 [32] for the carbohydrate atoms and with TIP3P for water molecules [33]. All calculations were run with the AMBER18 software package








Naphthalene diimide–amino acid conjugates as novel fluorimetric and CD probes for differentiation between ds-DNA and ds-RNA
Beilstein J. Org. Chem. 2020, 16, 2032–2045, doi:10.3762/bjoc.16.170

- with significant Stokes shifts (+60 nm) and quantum yields of 10–32% (Table 1). Theoretical calculations To get insight into the electronic and optical properties of the 2-amino-6-chloro-substituted NDIs, we carried out computational investigations by using the Gaussian 09 program suite [32]. In
- (solid line) and fluorescence spectra (dashed line) of NDI 3a,b, and 5 (c = 4.5 × 10−5 M in cacodylate buffer at pH 5.0, λex = 470 nm) at 23 °C. Calculations for Cl-NDI-NMe model compound (at the B3LYP/6-31+G** level of theory) in water (PCM). a) HOMO and LUMO molecular orbitals (isovalue surface 0.03 au
- vacuum. Bottom: The orientation of transition dipole moments (red arrow for 450–600 nm range) according to the calculations made for the spectra shown in Figure 2. Synthesis of water-soluble naphthalene diimides 3a,b, and 5. Optical properties of 3a,b, and 5 in cacodylate buffer (pH 5.0) at 23 °C. ∆Tm










A complementary approach to conjugated N-acyliminium formation through photoredox-catalyzed intermolecular radical addition to allenamides and allencarbamates
Beilstein J. Org. Chem. 2020, 16, 1983–1990, doi:10.3762/bjoc.16.165

- ]+); this peak persisted at 15, 30, 60 and 120 min time intervals, respectively. Upon the oxidation of 41 two plausible iminium stereoisomers can be formed, Z-42 and E-42, respectively, with each of these iminium stereoisomers existing in two further conformers designated E-42’ and Z-42’. DFT calculations






Natural dolomitic limestone-catalyzed synthesis of benzimidazoles, dihydropyrimidinones, and highly substituted pyridines under ultrasound irradiation
Beilstein J. Org. Chem. 2020, 16, 1881–1900, doi:10.3762/bjoc.16.156

- excellent AE (88–95%) and Curzon’s RME (78–93%) as well as a low to moderate E-factor (26.202–50.760) and PMI (27.202–51.760). The detailed calculations of the green chemistry metrics (AE, E-factor, PMI, Curzon’s RME, and gRME) for the synthesis of the compounds 3a, 7a, and 11a (Table 8, entry 1, Table 9











Synthesis and highly efficient light-induced rearrangements of diphenylmethylene(2-benzo[b]thienyl)fulgides and fulgimides
Beilstein J. Org. Chem. 2020, 16, 1820–1829, doi:10.3762/bjoc.16.149

- . The hydrogen atoms were localized in the difference-Fourier map and included in the refinement with fixed positional (riding model) and isotropic displacement parameters. All calculations were carried out using the SHELXTL program suite [26]. CCDC 1951883, 1951883 and 1951885 contain the supplementary














One-pot synthesis of oxazolidinones and five-membered cyclic carbonates from epoxides and chlorosulfonyl isocyanate: theoretical evidence for an asynchronous concerted pathway
Beilstein J. Org. Chem. 2020, 16, 1805–1819, doi:10.3762/bjoc.16.148
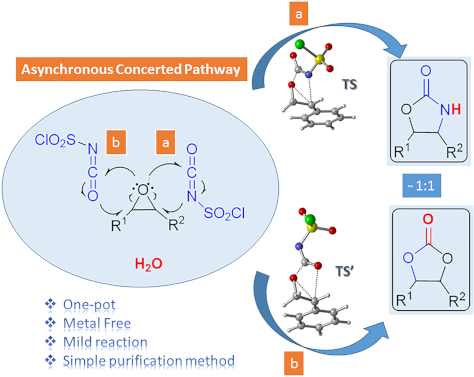
- reaction of CSI with epoxides in different solvents under mild conditions and compared the reaction mechanism with previously proposed mechanisms using theoretical calculations. Keshava Murthy and Dhar [41] postulated a mechanism involving a zwitterionic intermediate. C–O bond cleavage in this unstable and
- computational studies, such reactions of isocyanates may proceed through a concerted pathway. The remaining uncertainties in the mechanisms of the similar reactions inspired us to carry out quantum chemical calculations for the formation of oxazolidinone and five-membered cyclic carbonates. Results and
- isolated yields are 49% for 8f and 42% for 9f. The potential energy profiles of the formation of 8f and 9f are quite similar. IRC calculations revealed that the first step of the mechanisms for the formation of 8f and 9f occur asynchronously although in a concerted fashion. The water addition steps are














