Search results
Search for "computational" in Full Text gives 507 result(s) in Beilstein Journal of Organic Chemistry. Showing first 200.
Fluorinated azobenzenes as supramolecular halogen-bonding building blocks
Beilstein J. Org. Chem. 2019, 15, 2013–2019, doi:10.3762/bjoc.15.197
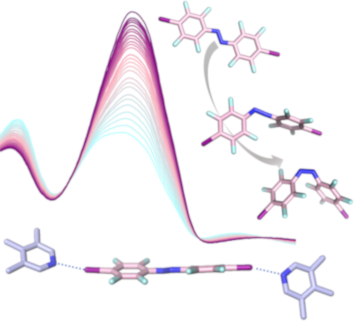
- * transitions of E and Z-isomers are listed in the Table S9 (Supporting Information File 1). The computational absorption spectra are in fair agreement with the experimental ones (Table 1) and trends are reproduced accordingly (measured and calculated absorption spectra of A1 can be found in the Supporting
- kinetic studies, computational methods, and X-ray crystallographic details. Acknowledgements We acknowledge the Fonds der Chemischen Industrie for a material cost allowance grant (B.M.S) and the Strategic Research Fund of Heinrich Heine University (F-2018/1460-4). E.N. is supported by a
- Deutschlandstipendium. We thank Y. Garmshausen for his advices on azobenzene photochemistry and C. Czekelius for sharing analytical equipment. Computational support and infrastructure was provided by the “Centre for Information and Media Technology” (ZIM) at the Heinrich Heine University.





Analysis of sesquiterpene hydrocarbons in grape berry exocarp (Vitis vinifera L.) using in vivo-labeling and comprehensive two-dimensional gas chromatography–mass spectrometry (GC×GC–MS)
Beilstein J. Org. Chem. 2019, 15, 1945–1961, doi:10.3762/bjoc.15.190

- and Tantillo, d9-α-ylangene with m/z = 213 should have been detected after in vivo labeling using [6,6,6-2H3]-(±)-mevalonolactone (d3-MVL) as precursor. At this point it should be mentioned that biosynthetic pathways in organisms can differ and computational studies only bear on the intrinsic




















Archangelolide: A sesquiterpene lactone with immunobiological potential from Laserpitium archangelica
Beilstein J. Org. Chem. 2019, 15, 1933–1944, doi:10.3762/bjoc.15.189
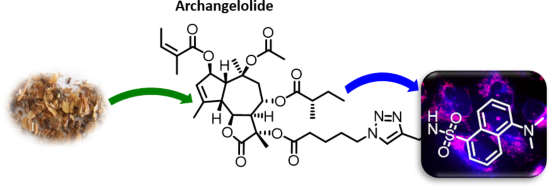
- pure product (37 mg, 0.04 mmol) as a slightly yellowish solid in 64% yield with Rf = 0.25 in hexanes/AcOEt, 1:1. For analytical data, see Supporting Information File 1, sections 2–4. Computational studies MD Simulations of compound 1 and 2 with sarco/endoplasmic reticular calcium ATPase For docking and
- ]; [LO1304] and by OP VaVpI project „Improving the quality of laboratory education of students at UCT Prague [KvaLab; 1.05/4.1.00/16.0349]. Computational resources were provided by the CESNET LM2015042 and the CERIT Scientific Cloud LM2015085, provided under the program "Projects of Large Research












Inherent atomic mobility changes in carbocation intermediates during the sesterterpene cyclization cascade
Beilstein J. Org. Chem. 2019, 15, 1890–1897, doi:10.3762/bjoc.15.184

- can sometimes bind to the active site in an unreactive conformation [11]. Recently, Siegel and Tantillo reported an innovative method for predicting the docking mode of carbocation intermediates in terpene cyclase [12][13], based on QM calculation and computational docking with the Rosetta protein
- software to calculate inherent atomic mobility. Acknowledgements This work was supported by JSPS KAKENHI (S) (No. 17H06173 (M.U.)), JSPS Grant-in-Aid for Scientific Research on Innovative Areas (No. 17H05430 (M.U.), JP16H06454 (M.Y.), and JP16H06443 (I.A.)). Provision of computational resources (Project







Halide metathesis in overdrive: mechanochemical synthesis of a heterometallic group 1 allyl complex
Beilstein J. Org. Chem. 2019, 15, 1856–1863, doi:10.3762/bjoc.15.181

- Information File 79: Experimental and computational details; crystal data and summary of X–ray data collection. Supporting Information File 80: Coordinates of DFT-optimized structures. Acknowledgements Financial support from the National Science Foundation (CHE-1665327) and the American Chemical Society




A golden opportunity: benzofuranone modifications of aurones and their influence on optical properties, toxicity, and potential as dyes
Beilstein J. Org. Chem. 2019, 15, 1781–1785, doi:10.3762/bjoc.15.171

- limited in scope to the influence of the benzylidene portion. Most noteworthy is the report by Bane and co-workers examining the UV–vis and fluorescent properties of a series of amino-substituted aurones [4]. Subsequently, Liu and co-workers explored the same series of aurones using computational methods
- . Muñoz-Becerra and co-workers have also reported a computational study of amino aurone derivatives with variations in the benzofuranone portion of the molecule, though all substitution was strictly at the 4-position of the benzofuranone [7]. As an extension of our on-going studies of the aurone ring





The cyclopropylcarbinyl route to γ-silyl carbocations
Beilstein J. Org. Chem. 2019, 15, 1769–1780, doi:10.3762/bjoc.15.170

- intermediates in these solvolysis reactions of 1 and 2. Labelling [13][14][15], stable ion [16][17][18][19], and computational studies [19] implicate the involvement of three degenerate cyclopropylcarbinyl cations, 6a, 6b, and 6c, in equilibrium with cyclobutyl cation 7, as well as the homoallylic cation 8
- cation rearrangement leads to the homoallylic products 40 and 41 via internal mesylate return or solvent capture. Of interest is the fact that no product 34 (derived from γ-trimethylsilyl-stabilized cation 37) is formed. Our previous computational study [52] provided insight into the lack of involvement
- . Computational studies support a carbocation intermediate with an unusually long Si–C bond, indicative of increased demand for Si–C hyperconjugation due to the electron-withdrawing group. With the exception of the phenyl substitution, the chemistry of trans-derivatives 14 is quite different since these





















Water inside β-cyclodextrin cavity: amount, stability and mechanism of binding
Beilstein J. Org. Chem. 2019, 15, 1592–1600, doi:10.3762/bjoc.15.163

- similar combined computational and experimental study of α-CD instead of β-CD hydration/dehydration has already been performed by us [19]. Results and Discussion Nonhydrated β-CD The nonhydrated β-CD molecules possess a nearly perfect 7-fold symmetry. The primary hydroxy groups (positioned at the upper
- flow calibration of the DSC was made by evaluating the melting peak of pure In and Zn. Dry nitrogen was used as purge gas at a fixed flow rate of 20 mL min−1. All DSC and DTA/TG measurements were repeated three times to ensure reproducibility and accuracy of the determined quantities. Computational
- experimental) approaches are sometimes conflicting and contradictory. There is no “standard” method to investigate the complexation behavior of CDs. Thus, in our study a computational quantum mechanical modelling method, namely density functional theory (DFT) was chosen to investigate the electronic structure







Transient and intermediate carbocations in ruthenium tetroxide oxidation of saturated rings
Beilstein J. Org. Chem. 2019, 15, 1552–1562, doi:10.3762/bjoc.15.158

- calculations [25] which were also in agreement with the earlier experiments of Bakke et al. [15]. The computational study also confirmed the hydroxide adduct Ib as the active intermediate formed in the reaction. However, Petride et al. have demonstrated that iminium cations are intermediates in the RuO4
- particular the analysis of the electron localization function (ELF) [39][40] is an excellent approach to evaluate the synchronicity of organic reactions [41][42] and consequently, to predict the formation of transient carbocations [43]. In this work, we report a computational study of the RuO4-mediated
- mechanism consisting of a (3 + 2) transition state has been confirmed as the preferred one [25], we restricted the study to this approach. Computational Methods The procedures are analogous to those previously reported [43]. All of the calculations were performed using the Gaussian 09 program [47












Superelectrophilic carbocations: preparation and reactions of a substrate with six ionizable groups
Beilstein J. Org. Chem. 2019, 15, 1515–1520, doi:10.3762/bjoc.15.153

- conversions, computational and experimental data indicated that the protonated hydroxy groups (oxonium ions) are not persistent intermediates, but rather cleavage of the carbon–oxygen bond is almost instantaneous [12]. It is assumed that ionization to the carbocations occurs in a stepwise process, first
- ]. Supporting Information Supporting Information File 484: Experimental procedures, compounds characterization, and NMR spectra; computational methods and results. Acknowledgements This work was supported by the National Science Foundation (1300878). We gratefully acknowledge this support.








Formation of an unexpected 3,3-diphenyl-3H-indazole through a facile intramolecular [2 + 3] cycloaddition of the diazo intermediate
Beilstein J. Org. Chem. 2019, 15, 1347–1354, doi:10.3762/bjoc.15.134

- structures stabilised by multiple tert-butyl groups [20][21], multi-ring cage hydrocarbons [22][23], and linear alkanes [22][23]. The interaction was found to be attractive in all these cases, and computational justifications have been published [21][23]. It, therefore, appears probable that, despite their










Host–guest interactions between p-sulfonatocalix[4]arene and p-sulfonatothiacalix[4]arene and group IA, IIA and f-block metal cations: a DFT/SMD study
Beilstein J. Org. Chem. 2019, 15, 1321–1330, doi:10.3762/bjoc.15.131

- shell of about 10), which is located outside the host cavity, has been found to be in agreement with the experimental data [32]. We report herein our computational (DFT) results on the complexation of p-sulfonatocalix[4]arene and thiacalix[4]arene with some metal guest cations. The thermodynamic
- solution of pH ≈ 2 the host calixarene systems have all the sulfonic acid groups deprotonated and all the phenolic hydroxy groups protonated thus the anionic structures shown on Scheme 1 were modeled and employed in our computational studies. Studies on the thermodynamic behavior and recognition processes
- thermodynamic parameters of the complex formation reaction meaningful. Computational The molecules of the ligands (calix[4]arenes and thiacalix[4]arenes), group IA, IIA and f-block metal cations and their complexes were optimized using the Gaussian 09 program package [38]. The computations were performed with








Mechanochemical Friedel–Crafts acylations
Beilstein J. Org. Chem. 2019, 15, 1313–1320, doi:10.3762/bjoc.15.130

- characterization data of compounds and computational procedures. Acknowledgements We acknowledge the financial support of the Ministry of Science, Education and Sport of Croatia (Project No. 098-0982933-2920).








Genomics-inspired discovery of massiliachelin, an agrochelin epimer from Massilia sp. NR 4-1
Beilstein J. Org. Chem. 2019, 15, 1298–1303, doi:10.3762/bjoc.15.128

- genome of this bacterium. Bioinformatic analyses greatly facilitated the stereochemical analysis and also demonstrated the usefulness of computational methods in the configurational assignment of this class of natural products. Experimental Analytical methods LC–MS analyses were performed with a



Bambusuril analogs based on alternating glycoluril and xylylene units
Beilstein J. Org. Chem. 2019, 15, 1268–1274, doi:10.3762/bjoc.15.124

- signals. X-ray crystal structure of 1b-1 (left), layered structure of crystal packing (right). Reaction of 2,4-dimethylglycoluril and m-xylylene dibromide. Supporting Information Supporting Information File 273: MS and NMR spectra, computational details and crystallographic data for macrocycles 1a and 1b







Precious metal-free molecular machines for solar thermal energy storage
Beilstein J. Org. Chem. 2019, 15, 1096–1106, doi:10.3762/bjoc.15.106

- spectra of the metal-free and Ba2+ complexed cis form of compound 4b are also presented in Figure 7. The oscillator strengths of the cis forms, calculated at the same computational level, are found to be significantly lower than those calculated for the respective trans forms (metal-free compound trans-4b
- fluorescent measurements was used allowing recording of the absorbance spectra during the laser irradiation in the perpendicular direction and immediately after the stop of irradiation. Computational details Equilibrium geometries and intermolecular interaction energies for the host–guest assemblies between











An anomalous addition of chlorosulfonyl isocyanate to a carbonyl group: the synthesis of ((3aS,7aR,E)-2-ethyl-3-oxo-2,3,3a,4,7,7a-hexahydro-1H-isoindol-1-ylidene)sulfamoyl chloride
Beilstein J. Org. Chem. 2019, 15, 931–936, doi:10.3762/bjoc.15.89

- reaction barriers for the formations of 10 and 11 is 3.6 kcal/mol, whereas that for the formations of 10 and 12 is 14.1 kcal/mol. Hence, the formation of 10 is kinetically more favorable. This computational result is consistent with the experimental observations. Conclusion For the first time, we have








Strong hyperconjugative interactions limit solvent and substituent influence on conformational equilibrium: the case of cis-2-halocyclohexylamines
Beilstein J. Org. Chem. 2019, 15, 818–829, doi:10.3762/bjoc.15.79

- National Center for High Performance Computing (CENAPAD-UFC) for the computational cluster disponibility.









Influence of per-O-sulfation upon the conformational behaviour of common furanosides
Beilstein J. Org. Chem. 2019, 15, 685–694, doi:10.3762/bjoc.15.63

- ++G** level using the COSMO continual solvation model with parameters for water. For complete computational details see the Experimental part. For all the three studied monosaccharides, both in non-sulfated (1–3) and sulfated (1s–3s) forms, the geometry optimizations tended to produce one or two low
- sheets coated with silica gel 60 F254 (Merck). Analysis TLC plates were developed by treatment with a mixture of 15% H3PO4 and orcinol (1.8 g/L) in EtOH/H2O (95:5, v/v) followed by heating. NMR and computational studies 1H and 13C NMR spectra were recorded on Bruker AV-400 or Bruker Fourier 300HD
- . Experimental 1H–1H coupling constants (Hz) for α-propyl mannofuranosides (1, 1s) and those calculated for its different conformers (Hz). Supporting Information Supporting Information File 118: Copies of 1H and 13C NMR spectra of compounds 1–3 and 1s–3s and computational details for all found conformers





Catalyst-free assembly of giant tris(heteroaryl)methanes: synthesis of novel pharmacophoric triads and model sterically crowded tris(heteroaryl/aryl)methyl cation salts
Beilstein J. Org. Chem. 2019, 15, 642–654, doi:10.3762/bjoc.15.60

- over MgSO4, and concentrated under reduced pressure. Finally, the crystals were obtained after simple trituration with Et2O. Computational methods. Density functional theory (DFT) calculations were carried out with the Gaussian 09 program suite [66]. Geometries were fully optimized at the B3LYP [67][68

















Back to the future: Why we need enzymology to build a synthetic metabolism of the future
Beilstein J. Org. Chem. 2019, 15, 551–557, doi:10.3762/bjoc.15.49

- can these new enzyme reactions be identified or established? One option is the de novo-design of enzymes assisted by computational methods, which have been developed over the last couple of years. When combined with experimental evolution and elaborate screening methods, these efforts have allowed to
- establish completely novel enzyme reactions from scratch [31][32][33][34]. However, even though considerable progress has been made in creating enzymes with the help of computational methods [35], it is a complementary (and equally valid) approach to discover and/or engineer novel reactions from the natural
- methylsuccinyl-CoA oxidase in the CETCH cycles that were engineered from a promiscuous short chain acyl-CoA oxidase and a FAD-dependent methylsuccinyl-CoA dehydrogenase, respectively [13][40][41]. These efforts in exploiting the promiscuity of enzymes to create novel catalysts might profit from new computational



Design of indole- and MCR-based macrocycles as p53-MDM2 antagonists
Beilstein J. Org. Chem. 2019, 15, 513–520, doi:10.3762/bjoc.15.45

- HSQC NMR spectra of 15N-labeled MDM2 and computational modeling studies. Acknowledgements This research has been supported to (AD) by the National Institute of Health (NIH) (2R01GM097082-05), the European Lead Factory (IMI) under grant agreement number 115489, the Qatar National Research Foundation






Conformational signature of Ishikawa´s reagent using NMR information from diastereotopic fluorines
Beilstein J. Org. Chem. 2019, 15, 506–512, doi:10.3762/bjoc.15.44

- provided detailed account on the H–C2–C1–F dihedral angle as the solvent varied. Experimental and Computational Details N,N-Diethyl-(1,1,2,3,3,3-hexafluoropropyl)amine (1) was commercially available (90% purity) and used without further purification. The NMR spectra were acquired at 400.2 or 499.9 MHz for



Study on the regioselectivity of the N-ethylation reaction of N-benzyl-4-oxo-1,4-dihydroquinoline-3-carboxamide
Beilstein J. Org. Chem. 2019, 15, 388–400, doi:10.3762/bjoc.15.35

- crystallographic table was mounted using the OLEX2 software [36]. Computational details All the calculations were carried out with the Gaussian 09 software package [37] considering the absence (gas phase) and the presence of two implicit solvents (water and DMSO), using the polarized continuum solvation model












Sigmatropic rearrangements of cyclopropenylcarbinol derivatives. Access to diversely substituted alkylidenecyclopropanes
Beilstein J. Org. Chem. 2019, 15, 333–350, doi:10.3762/bjoc.15.29

- ’ would then be obtained and would eventually produce the diastereomeric phosphine oxides 7 and 7’. Computational studies indicated that the facial selectivity of the initial attack of the Lewis base (DBU) was not responsible for the observed diastereocontrol because of the low difference between the



























