Search results
Search for "DFT" in Full Text gives 534 result(s) in Beilstein Journal of Organic Chemistry. Showing first 200.
Synthesis of purines and adenines containing the hexafluoroisopropyl group
Beilstein J. Org. Chem. 2020, 16, 2739–2748, doi:10.3762/bjoc.16.224

- 3a at 253 K. Right: Eyring plot of the interconversion of the rotamers of 3a. DFT-optimized structures of the two rotamers of 3a. Left: Lower-energy rotamer. Right: Higher-energy rotamer. Reaction of purine (2) with tetrakis(trifluoromethyl)-1,3-dithietane (1). Reaction of 4-azabenzimidazole (3) with











Vicinal difluorination as a C=C surrogate: an analog of piperine with enhanced solubility, photostability, and acetylcholinesterase inhibitory activity
Beilstein J. Org. Chem. 2020, 16, 2663–2670, doi:10.3762/bjoc.16.216

- . This was achieved by performing a DFT study in the Gaussian software, using the M06-2X level of theory with the 6-311+G(d,p) basis set, parameters similar to those employed by Linclau and co-workers for their studies of vicinal difluoro systems [22]. A set of starting structures of 2 was generated by
- physicochemical properties of compound 2 were found to be superior to piperine (1) itself in two key respects, namely photostability and aqueous solubility. The conformational analysis of 2 by DFT and NMR spectroscopy revealed that the lowest-energy conformations 2a–c are imperfect mimics of 1 but that a somewhat
- for this work; the crystal structure is shown [14]. In this work, a hypothetical analog 2 was designed to mimic parent compound 1. The predicted low-energy rotamers of 2 about the F–C–C–F and F–C–C=O bonds are shown; rotamers I and IV give the best mimicry of 1. Conformational analysis of 2 by DFT and








Particle size effect in the mechanically assisted synthesis of β-cyclodextrin mesitylene sulfonate
Beilstein J. Org. Chem. 2020, 16, 2598–2606, doi:10.3762/bjoc.16.211

- defined as the ratio of the micropore volume to the total pore volume. The relative errors were estimated to be the following: SBET, 5%; pore volume (pv, DFT), 5%; pore size (ps, DFT), 20%. Isotherms were measured on a Quantachrome® ASiQwin™ instrument at 0 and 21 °C. The temperature was held constant









Synthesis of 4-substituted azopyridine-functionalized Ni(II)-porphyrins as molecular spin switches
Beilstein J. Org. Chem. 2020, 16, 2589–2597, doi:10.3762/bjoc.16.210

- is always in a high paramagnetic state (>80%) and showed only minor switching efficiency after irradiation (4%). This is attributed to intramolecular coordination of the trans state (Supporting Information File 1, Figure S17). Application of density functional theory (DFT) at the B3LYP/def2TZVP//PBE









Water-soluble host–guest complexes between fullerenes and a sugar-functionalized tribenzotriquinacene assembling to microspheres
Beilstein J. Org. Chem. 2020, 16, 2551–2561, doi:10.3762/bjoc.16.207

- Ka = (2.20 ± 0.16) × 105 M−1, respectively. The binding affinity between TBTQ-(OG)6 and C60 was further verified by Raman spectroscopy. The geometry of the complex of TBTQ-(OG)6 with C60 deduced from DFT calculations indicates that the driving force of the complexation is mainly due to the
- diffraction. Therefore, the optimized geometry of the 1:1 complex of TBTQ-(OG)6 C60 in water was simulated by density functional theory (DFT) calculations at the B3LYP/6-31G(d) level of theory, which was completed with the aid of Molclus, MOPAC, and ORCA 4.1.0 programs [54][55][56]. As shown in Figure 6, C60
- were dried on a slide glass. C60 was tested in powder form on a slide glass. Molecular model of the complex TBTQ-(OG)6 C60 in water, as generated by DFT calculations. (a) Side-view; (b) top-view; (c) hydrophobic surface diagram. In part, H atoms were omitted for clarity (yellow: C, red: O, blue: N





![[Graphic 2]](/bjoc/content/inline/1860-5397-16-207-i3.svg?max-width=637&scale=1.18182) C60 [TBTQ-(OG)6: 50 μM; C60: 50 μM] and (b) TBTQ-(OG)6
C60 [TBTQ-(OG)6: 50 μM; C60: 50 μM] and (b) TBTQ-(OG)6 



Conformational preferences of fluorine-containing agrochemicals and their implications for lipophilicity prediction
Beilstein J. Org. Chem. 2020, 16, 2469–2476, doi:10.3762/bjoc.16.200

- high degree of freedom in the chemical structure of penoxsulam (I), the conformational analysis started with a Monte Carlo conformational search at the ωB97X-D/6-31G(d,p) [16][17] level of the density functional theory (DFT). The global energy minimum conformation was then re-optimized in a higher








Recent developments in enantioselective photocatalysis
Beilstein J. Org. Chem. 2020, 16, 2363–2441, doi:10.3762/bjoc.16.197
















































































Styryl-based new organic chromophores bearing free amino and azomethine groups: synthesis, photophysical, NLO, and thermal properties
Beilstein J. Org. Chem. 2020, 16, 2282–2296, doi:10.3762/bjoc.16.189

- dyes bearing the julolidine donor as 1430 × 10−48 esu (for free amino derivative) and 1950 × 10−48 esu (for Schiff base derivative), respectively. The structural and electronic properties of the dyes as well as their NLO properties were further studied using DFT calculations. The thermal stabilities of
- all dyes were evaluated by thermogravimetric analysis (TGA). The TGA data showed that all dyes were thermally stable up to 250 °C. Keywords: DFT calculations; NLO; pH sensitive dyes; Schiff base; solvent effect; styryl dyes; Introduction Push-pull organic molecules are a class of organic dyes
- series of new styryl-based organic chromophores containing a free amino group and the corresponding Schiff base derivatives. The photophysical, pH sensitivity, NLO properties, and thermal stabilities of all synthesized dyes were investigated. Density Functional Theory (DFT) calculations were also












Synthetic approaches to bowl-shaped π-conjugated sumanene and its congeners
Beilstein J. Org. Chem. 2020, 16, 2212–2259, doi:10.3762/bjoc.16.186

- thermoelectric properties in addition to the onigiri-type core-shell assemblies have been reported for sumanene and its derivatives. More interestingly, its application in the absorption of small molecules such as NH3, CO2, CO, and H2 using density functional theory (DFT) calculations has also been revealed [25
- /mol) transformation of 12 (syn) to 17, calculated by density functional theory (DFT) calculations. On the other hand, in 2008, Higashibayashi et al. reported the synthesis of first chiral C3-symmetric trimethylsumanene 28 starting from enantiopure norbornadiene (10) by employing a rational synthetic
- (DFT) calculations. As we are aware that if the carbene formed is a singlet then a C–H bond inserted product is predominating whereas if the dimer is the major product along with the minor C–H bond inserted product then the triplet carbene is generated. During their study, they obtained the C–H






































































Photosensitized direct C–H fluorination and trifluoromethylation in organic synthesis
Beilstein J. Org. Chem. 2020, 16, 2151–2192, doi:10.3762/bjoc.16.183

- TA spectra. The direct reaction between AQN and the substrate was not observed by TAS. The authors’ DFT calculations revealed reaction pathways that were thermodynamically and kinetically plausible. Initially, AQN and Selectfluor® (S0 in Scheme 16) form a van der Waals complex RC1, which is markedly






















































Lipophilicity trends upon fluorination of isopropyl, cyclopropyl and 3-oxetanyl groups
Beilstein J. Org. Chem. 2020, 16, 2141–2150, doi:10.3762/bjoc.16.182

- even for a set of relatively simple compounds, fragment-based clogP calculations are not guaranteed to give reliable lipophilicity data. Next, theoretical lipophilicities were obtained using DFT calculations, based on the notion that the partition coefficient of a given solute between two phases
- provide any useful approximation in terms of rank order or absolute magnitude of effect. For series D, E, G, the DFT-logP values cluster together with minimal lipophilicity differences, and within mean absolute error limits (estimated to be around 0.8 logP units), making detailed considerations
- meaningless. This was a surprise, as series D and E are relatively rigid, which simplifies conformational analysis. The remarkably similar DFT-logP values suggest that the influence of the fluorination is underrepresented in the calculations. The following observations are noteworthy. The trifluorinated D5












Muyocopronones A and B: azaphilones from the endophytic fungus Muyocopron laterale
Beilstein J. Org. Chem. 2020, 16, 2100–2107, doi:10.3762/bjoc.16.177

- ) [25]. After conformational analysis, geometry optimization was performed for two possible stereoisomers with the (7S,10R,11R)- and (7S,10S,11S)-configurations using density functional theory (DFT) at the CAM-B3LYP/6-311+G(d,p) level of theory. In addition, the ECD spectra of the DFT-optimized
- conformers were calculated using time-dependent DFT (TDDFT) at the B3LYP/6-311+G(d,p) level of theory. The negative Cotton effect observed at 236 nm in the measured spectrum was in good agreement with that in the calculated ECD spectrum of the (7S,10R,11R)-stereoisomer (Figure 4A). Thus, the absolute
- –7.62 (m, 10H, phenyl groups of MTPAs). Computational methods Calculation of the ECD spectra were performed using CONFLEX 8, Gaussian 16, and SpecDis software as described previously [13][14]. Geometry optimizations were performed using DFT at the CAM-B3LYP/6-311+G(d,p) level of theory, while TDDFT






Naphthalene diimide–amino acid conjugates as novel fluorimetric and CD probes for differentiation between ds-DNA and ds-RNA
Beilstein J. Org. Chem. 2020, 16, 2032–2045, doi:10.3762/bjoc.16.170

- substituents in the imide positions and on the nitrogen on the core are known to have a negligible effect on the chromophore, since both the HOMO and the LUMO have a node at these positions [33]. Geometry optimization by DFT method (at the B3LYP/6-31+G** level of theory) confirmed a planar and rigid NDI
- molecular ground-state electrical dipole (µ = 7.04 D), which is an evidence for an intramolecular charge transfer (CT) character of the chromophore [34]. The predicted UV–vis spectra by TD-DFT are in excellent agreement with the experimental one (Figure 3, Table 1, the vibrational coupling is neglected
- ). b) The molecular ground state dipole moment is indicated by the black arrow. The transition dipole moments calculated by TD-DFT (at the B3LYP/6-31+G** level of theory) for the lowest transitions in the visible range of the spectrum (indicated in the spectra by the red and blue arrows) are shown










A complementary approach to conjugated N-acyliminium formation through photoredox-catalyzed intermolecular radical addition to allenamides and allencarbamates
Beilstein J. Org. Chem. 2020, 16, 1983–1990, doi:10.3762/bjoc.16.165

- findings on the performance of allenamides. Specifically, the electron-withdrawing group on the allenamide and the nucleophile is examined. We provide evidence for the formation of the N-acyliminium intermediate through direct sample loop and flow injection analysis by HRESIMS, and DFT analysis of the N
- ]+); this peak persisted at 15, 30, 60 and 120 min time intervals, respectively. Upon the oxidation of 41 two plausible iminium stereoisomers can be formed, Z-42 and E-42, respectively, with each of these iminium stereoisomers existing in two further conformers designated E-42’ and Z-42’. DFT calculations






Synthesis, docking study and biological evaluation of ᴅ-fructofuranosyl and ᴅ-tagatofuranosyl sulfones as potential inhibitors of the mycobacterial galactan synthesis targeting the galactofuranosyltransferase GlfT2
Beilstein J. Org. Chem. 2020, 16, 1853–1862, doi:10.3762/bjoc.16.152

- mechanism studies using computational chemistry methods. The probable reaction mechanisms were studied by hybrid DFT QM/MM molecular dynamics simulations [11] where the possible transition state (TS) structures were localized. The observation of the possible TS structure opens the opportunities for the in










One-pot synthesis of oxazolidinones and five-membered cyclic carbonates from epoxides and chlorosulfonyl isocyanate: theoretical evidence for an asynchronous concerted pathway
Beilstein J. Org. Chem. 2020, 16, 1805–1819, doi:10.3762/bjoc.16.148
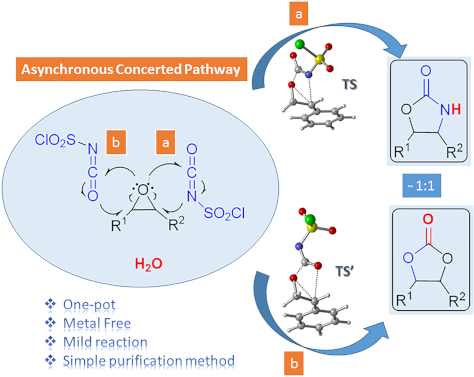
- advantageous: being a one-pot reaction with metal-free reagent, having shorter reaction times, good yields and a very simple purification method. Moreover, using the density functional theory (DFT) method at the M06-2X/6-31+G(d,p) level of theory the mechanism of the cycloaddition reactions has been elucidated















Pauson–Khand reaction of fluorinated compounds
Beilstein J. Org. Chem. 2020, 16, 1662–1682, doi:10.3762/bjoc.16.138

- substituents such as dimethylaminium, trifluoromethyl, and acetyl favored the β-regioisomer (71B). The 4-fluorine substituted diarylalkynes had a very weak EWG effect yielding an equimolar mixture of both regioisomers. The experimental results were confirmed by a DFT study of the NBO charges of the α-alkyne












































Rearrangement of o-(pivaloylaminomethyl)benzaldehydes: an experimental and computational study
Beilstein J. Org. Chem. 2020, 16, 1636–1648, doi:10.3762/bjoc.16.136

- )benzaldehydes under acidic conditions resulted in the formation of the regioisomeric aldehydes and/or dimer-like products. Detailed NMR studies and single-crystal X-ray measurements supported the structure elucidation of the compounds. DFT calculations were also carried out to clarify the reaction mechanism
- , and to explain the observed product distributions and structural variances in the dimer-like products. Studies on the transformation of unsubstituted o-(pivaloylaminomethyl)benzaldehyde under similar conditions were presented as well. Keywords: DFT calculations; NMR; reaction mechanism; rearrangement
- reaction described in Scheme 1 to further o-(pivaloylaminomethyl)benzaldehydes and to support the results by density functional theory (DFT) calculations, single-crystal X-ray measurements and comprehensive NMR studies. Results and Discussion Acid-catalyzed transformations of compounds 1a–d First we kept













Heterogeneous photocatalysis in flow chemical reactors
Beilstein J. Org. Chem. 2020, 16, 1495–1549, doi:10.3762/bjoc.16.125

- measurements, TRPL spectroscopy, and density functional theory (DFT) calculations. They found that the HER efficiency correlated to the excited state lifetime and exciton binding energy. The FSO-BP and FSO-FSz hindered the charge transfer and mobility due to the phenyl–phenyl dihedral angle or sharp bends in

















































In silico rationalisation of selectivity and reactivity in Pd-catalysed C–H activation reactions
Beilstein J. Org. Chem. 2020, 16, 1465–1475, doi:10.3762/bjoc.16.122
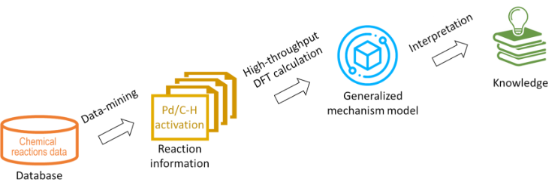
- the mechanistic models based on accurate quantum chemical methods, such as the density functional theory (DFT) methods, decreases. Automation of DFT, as well as using results of DFT to develop less expensive predictive models, are the two approaches that may offer the alternatives to the fully data
- -driven statistical methods. Here we demonstrate an approach that was developed to automate the DFT-level calculations of energies of the auto-generated reaction intermediates. These results were further used to generalize mechanistic knowledge of a class of reactions, and the developed models were used
- transition metal coordination sphere; the energy of a new M–C bond formed and the thermodynamic stability of organometallic product. With new developments in computational chemistry, mechanistic studies using density functional theory (DFT) provide valuable insights into the reactivity of organometallic



Development of fluorinated benzils and bisbenzils as room-temperature phosphorescent molecules
Beilstein J. Org. Chem. 2020, 16, 1154–1162, doi:10.3762/bjoc.16.102

- -deficient fluorinated aromatic ring. To confirm the electron-withdrawing effect of this fluorinated aromatic ring, the electronic charge at the adjacent C≡C bond was calculated by density functional theory (DFT) using the Gaussian 16 (Revision B.01) software package [35]. As typical examples, the molecular
- –405 nm (ε: 180–260 M−1·cm−1). To gain more information about the slight difference between the absorption behaviors of the benzil and bisbenzil derivatives, DFT and time-dependent DFT (TD-DFT) calculations at the CAM-B3LYP/6-31+G(d) level of theory were performed for fluorinated benzil 2a and
- lobe is localized at the central benzene ring. Accordingly, it can be concluded that the absorption bands at the short-wavelength region (around 290–315 nm) stems from the π–π* transition. The TD-DFT calculation also reveals that n–π* transitions (e.g., HOMO–2→LUMO for 2a and HOMO–3→LUMO for 3a) have








Synthesis and properties of quinazoline-based versatile exciplex-forming compounds
Beilstein J. Org. Chem. 2020, 16, 1142–1153, doi:10.3762/bjoc.16.101

- [15]. Theoretical calculations and electrochemical properties DFT calculations were employed to gain insight into the structure–property relationships of the quinazoline-based derivatives 1–3. The compounds have phenyl spacers between the donor and acceptor units (Figure 2). Therefore, the dihedral
- energy values at the corresponding excited-state geometry. The time-dependent DFT (TD-DFT) calculations were carried out with the Gaussian 16 software package and molecular orbitals were visualized by using Gaussview. Photoelectron emission spectroscopy measurement was performed according to the









A cyclopeptide and three oligomycin-class polyketides produced by an underexplored actinomycete of the genus Pseudosporangium
Beilstein J. Org. Chem. 2020, 16, 1100–1110, doi:10.3762/bjoc.16.97

- -acetyl-ʟ-Tyr-ʟ-Pro-ʟ-Trp, was determined by a combination of spectroscopic analyses, chemical derivatization, ECD calculation, and DFT-based theoretical chemical shift calculation, revealing the presence of an (Sa)-axial chirality around the biaryl bond. Compounds 2–4 lacked hydroxylation on the side
- discovery of new natural products. Keywords: DFT-based calculation; oligomycin; peptide; polyketides; Pseudosporangium; rare actinomycetes; Introduction Microbial secondary metabolites have been used as therapeutic drugs [1], veterinary medicines [2], agrochemicals [3], food preservatives/colorings [4][5
- comparison of experimental and simulated ECD spectra [26][27]. Quite interestingly, the DFT calculation suggested that 1a and 1b would possess the opposite axial chirality around the biaryl bond between C-6 and C-7'': Ra for 1a and Sa for 1b as dominant atropisomers (Figure 4). This conformational difference









Aryl-substituted acridanes as hosts for TADF-based OLEDs
Beilstein J. Org. Chem. 2020, 16, 989–1000, doi:10.3762/bjoc.16.88

- . Theoretical calculations The optimized structures of 3–6 were obtained by density functional theory (DFT) calculations at the B3LYP/6-31G(d,p) level of theory (Figure 1). The dihedral angles between the acridanyl and phenyl moieties in compound 3 (37.0 and 36.3°) are comparable with the dihedral angles
- , 29.3, 11.7; MS (APCI+, 20 V) m/z: 426 ([M + H]+); anal. calcd for C29H25F2N: C, 81.86; H, 5.92; F, 8.93; N, 3.29; found: C, 81.91; H, 5.99; N, 3.31%. Theoretically calculated HOMO and LUMO levels distributions and optimized geometries of 3–6 DFT calculations were performed at the B3LYP/6-31G(d,p) level








Synthesis and properties of tetrathiafulvalenes bearing 6-aryl-1,4-dithiafulvenes
Beilstein J. Org. Chem. 2020, 16, 974–981, doi:10.3762/bjoc.16.86

- ; one was the palladium-catalyzed C–H arylation of TTF with bromide 12 (Scheme 2a) and the other was the Vilsmeier–Haack reaction of 1a, followed by triethyl phosphite-mediated cross coupling with 11 (Scheme 2b). Theoretical calculations The DFT calculations of compounds 1a, 3a, and 4 have been carried
- is participating in this redox process is surrounded by extended aromatic rings bearing 1,3-dithiol rings. Conclusion We have synthesized novel multistage TTF derivatives 1–4 bearing 6-aryl-1,4-dithiafulvene moieties by palladium-catalyzed direct C–H arylation. The DFT calculations revealed the







