Search results
Search for "electron-transfer" in Full Text gives 287 result(s) in Beilstein Journal of Organic Chemistry. Showing first 200.
Non-noble metal-catalyzed cross-dehydrogenation coupling (CDC) involving ether α-C(sp3)–H to construct C–C bonds
Beilstein J. Org. Chem. 2023, 19, 1259–1288, doi:10.3762/bjoc.19.94

- bonds The possible mechanism of the CDC reaction involving ether α-C(sp3)–H bonds mainly follows the two pathways outlined in Scheme 2. Route a: First, the C(sp3)–H bond at the α-position of the oxygen atom undergoes a single-electron transfer under the combined action of the transition metal and an
- extract a hydrogen from the ether C (sp3)–H bond to form radicals. Subsequently, a single electron transfer (SET) leads to the oxonium species. Then, the enamine generated in situ from methyl aryl ketone and pyrrolidine undergoes a nucleophilic reaction with the oxonium species followed by hydrolysis to
- of the radical initiator to the ether to obtain the corresponding ether radical species. The coupling product is accessed through a single electron transfer (SET) and other transformations. In 2019, Tu et al. established a highly efficient Cu-catalyzed cross-dehydrogenative coupling to access a












































Radical ligand transfer: a general strategy for radical functionalization
Beilstein J. Org. Chem. 2023, 19, 1225–1233, doi:10.3762/bjoc.19.90

- Fe(II/III) cycle is proposed, where a single electron transfer from Fe(II) reduces the peroxyester and produces a carboxyl radical and Fe(III), which can coordinate an azide ligand. Rapid decarboxylation produces the transient alkyl radical which can be asymmetrically azidated by RLT from an Fe(III
- the addition of terminal oxidant. II: The proposed mechanism includes reoxidation of the iron catalyst through inner-sphere electron transfer by anionic nitrate. Funding J.G.W. acknowledges financial support from CPRIT (RR190025), NIH NIGMS (R35GM142738), Research Corporation for Science Advancement






Enabling artificial photosynthesis systems with molecular recycling: A review of photo- and electrochemical methods for regenerating organic sacrificial electron donors
Beilstein J. Org. Chem. 2023, 19, 1198–1215, doi:10.3762/bjoc.19.88
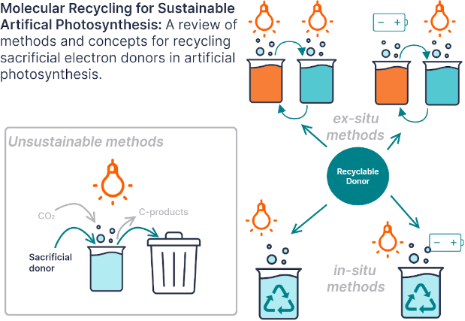
- quenched by a substrate or catalyst and then reduced by the sacrificial donor (oxidative quenching pathway). In the presence of protons, proton donors, or oxidized donor species with a low pKa, a proton-coupled electron transfer (PCET) can take place [14][15]. PCET reactions are important in artificial
- or oxidized photosensitizer is the driving force for the electron transfer and photosensitizer regeneration. This driving force determines the rate of electron transfer from the electron donor to the photosensitizer which regulates the amount of photosensitizer available to harvest light energy and
- and allows the accumulation of the oxidized species and almost complete consumption of the sacrificial donor. However, thermodynamically irreversible does not mean that the sacrificial donor must break down into unrecyclable fragments like triethylamine (TEA). An irreversible electron transfer is slow






Exploring the role of halogen bonding in iodonium ylides: insights into unexpected reactivity and reaction control
Beilstein J. Org. Chem. 2023, 19, 1171–1190, doi:10.3762/bjoc.19.86

- cycloaddition reactions that occur without transition metal catalysts, the unexpected initiation of single electron transfer (SET) processes or photochemical transformations, and even proton transfers that appear to defy pKa limitations. The reaction pathways followed by iodonium ylides and Lewis basic reaction
- , they believed that the reaction was likely initiated by either single electron transfer between the reagents (not shown), or by electrophilic addition of the olefin onto the ylide, forming intermediate adduct 17. This was followed by formation of iodocycle 18, from which reductive elimination of
- as an electron donor–acceptor complex) [121], and this bonding description has recently been used to support proposals for single electron transfer (SET) reaction pathways between iodonium ylides and various halogen bond acceptors. Alternatively, halogen-bonded complexes of iodonium ylides could lead



































Selective and scalable oxygenation of heteroatoms using the elements of nature: air, water, and light
Beilstein J. Org. Chem. 2023, 19, 1146–1154, doi:10.3762/bjoc.19.82

- separate” additives, a significant rate enhancement could be obtained with a positive impact on productivity rates. Results and Discussion There are a lot of similarities between electrochemistry and photoredox chemistry [33] as both rely on single-electron transfer processes to initiate reactions. In
- electrochemistry, the electron transfer occurs locally at the surface of the physical electrodes (typically located at a distance in the range of 200 μm to 2 cm) on which a potential is induced by an external potentiostat (Scheme 2). While for photoredox chemistry, the light-activated semiconductor catalyst
- irradiation at 310 nm [27]. They concluded that both a single-electron transfer and a singlet oxygen path can occur depending on the nature of the compound. Intriguingly, the current method applies 365 nm irradiation at which thioanisole does not absorb (Supporting Information File 1, Figure S1). Also, the







Photoredox catalysis harvesting multiple photon or electrochemical energies
Beilstein J. Org. Chem. 2023, 19, 1055–1145, doi:10.3762/bjoc.19.81

- Mattia Lepori Simon Schmid Joshua P. Barham Fakultät für Chemie und Pharmazie, Universität Regensburg, Universitatsstraße 31, 93040 Regensburg, Germany 10.3762/bjoc.19.81 Abstract Photoredox catalysis (PRC) is a cutting-edge frontier for single electron-transfer (SET) reactions, enabling the
- , excellent alternative conditions are available to overcome these limitations, harvesting two different but correlated concepts: the use of multi-photon processes such as consecutive photoinduced electron transfer (conPET) and the combination of photo- and electrochemistry in synthetic photoelectrochemistry
- the most appropriate for a given reaction, scale and purpose of a project. Keywords: consecutive photoinduced electron transfer; electro-activated photoredox catalysis; photoelectrochemistry; photoredox catalysis; radical ions; Review 1 Introduction Owing to the unique reactivity patterns of free













































































Photoredox catalysis enabling decarboxylative radical cyclization of γ,γ-dimethylallyltryptophan (DMAT) derivatives: formal synthesis of 6,7-secoagroclavine
Beilstein J. Org. Chem. 2023, 19, 918–927, doi:10.3762/bjoc.19.70

- manner due to their intrinsic mildness and broad substrate compatibility [16][17][18][19][20]. This transformative synthetic tool often utilizes direct single-electron transfer (SET) between an electronically excited photoredox catalyst and an organic substrate, resulting in oxidation or reduction, to
- position via a sequential electron transfer–proton transfer (ET/PT) [52][53][54][55][56][57][58][59]. With our ongoing interest of establishing new methods for the asymmetric synthesis of nonproteinogenic tryptophan derivatives as well as their associated indole alkaloid natural products [60][61][62][63








Intermediates and shunt products of massiliachelin biosynthesis in Massilia sp. NR 4-1
Beilstein J. Org. Chem. 2023, 19, 909–917, doi:10.3762/bjoc.19.69

- photosynthesis, respiration or nitrogen fixation, in which iron-containing proteins are engaged in electron transfer reactions. In fact, the transition metal is perfectly suited for shifting electrons due to its ability to easily interconvert between a reduced ferrous (Fe2+) and an oxidized ferric state (Fe3





Sulfate radical anion-induced benzylic oxidation of N-(arylsulfonyl)benzylamines to N-arylsulfonylimines
Beilstein J. Org. Chem. 2023, 19, 771–777, doi:10.3762/bjoc.19.57

- single electron transfer (SET), is proposed to be involved in the plausible reaction mechanism. Keywords: arylsulfonylimine; benzylic oxidation; benzyl sulfonamide; K2S2O8; sulfate radical anion; Introduction Among various imine compounds [1], N-arylsulfonylimines are perhaps the most prominent due to
- abstraction (HAT) followed by single electron transfer (SET) enabled by the sulfate radical anion (SO4·−). Results and Discussion Initially, we investigated the reaction of N-benzenesulfonyl(benzyl)amine (1a) as a model substrate with K2S2O8 in MeCN at 80 °C for 12 h, conditions that were used earlier in our
- cleavage of the peroxy linkage under heating conditions [17]. The hydrogen atom is abstracted from the benzylic position of 1 by SO4·−, generating benzylic radical 1aa [14][15][16]. A single electron transfer (SET) could subsequently occur from 1aa to form the reactive species 1ab. Finally, the base







Synthesis, structure, and properties of switchable cross-conjugated 1,4-diaryl-1,3-butadiynes based on 1,8-bis(dimethylamino)naphthalene
Beilstein J. Org. Chem. 2023, 19, 674–686, doi:10.3762/bjoc.19.49

- , electron transfer, and quantum interference [17][18][19][20], but are also considered as promising molecular switches and transistors [21][22][23][24][25], NLO materials [26][27][28][29], and suitable starting compounds for syntheses involving multiple Diels–Alder additions [30]. All these facts motivated
- monomers 6d and 6e are typical rod-like D–π–A systems. In such molecules, a photon absorption induces a shift from a D–π–A ground state to a D+–π–A− excited state. It is obvious that the actual electron transfer depends on all three components of the push–pull molecule. However, the study on the through
- and 6b·HBF4. Since the UV–vis spectra of salts 11 are similar (even and especially in cases of methoxy and nitro derivatives 11e and 11b), it can be assumed that in all salts 11 the electron transfer from the terminal aryl to the central naphthalene rings takes place. Naturally, the more deficient the

















Direct C2–H alkylation of indoles driven by the photochemical activity of halogen-bonded complexes
Beilstein J. Org. Chem. 2023, 19, 575–581, doi:10.3762/bjoc.19.42

- charge transfer state which results in a bathochromic shift of the absorption towards the visible range [19][20]. Upon light irradiation, the EDA complex may undergo an intramolecular single-electron-transfer (SET) process to produce a radical ion pair (D•+, A•−). To avoid the occurrence of a back
- -electron-transfer (BET), a suitable leaving group (LG) needs to be included in one of the precursors. In this manner, reactive intermediates (e.g., radical species) may be generated in solution through the irreversible fragmentation of the substrates [15][21][22]. These intermediates eventually react to






A new oxidatively stable ligand for the chiral functionalization of amino acids in Ni(II)–Schiff base complexes
Beilstein J. Org. Chem. 2023, 19, 566–574, doi:10.3762/bjoc.19.41

- fragment in the oxidized (GlyNi)L7 and (ΔAlaNi)L7 complexes bears a certain spin density (as it has been previously shown for the t-Bu-free analogs [31]), the bulky t-Bu substituent does prevent dimerization of the radical cations formed in the electron-transfer step increasing their kinetic stability
- reversibility can be reached at scan rates above 20 V/s allowing the E1/2 estimation. Similarly to previously studied complex (ΔAlaNi)L1 [36], electron transfer to (ΔAlaNi)L7 is mainly ligand centered. The DFT-estimated LUMO is formed as an overlap of the π antibonding orbitals of the dehydroalanine and imine







CuAAC-inspired synthesis of 1,2,3-triazole-bridged porphyrin conjugates: an overview
Beilstein J. Org. Chem. 2023, 19, 349–379, doi:10.3762/bjoc.19.29

- -dipolar cycloaddition reaction between an azide and a terminal alkyne, also popular as "click reaction" or CuAAC reaction. Moreover, the 1,2,3-triazole ring also serves as a spacer and an electron transfer bridge between the porphyrin and the attached chromophores. In order to provide a critical overview
- a photophysical investigation, a weak interaction was observed between two subunits at ground state in porphyrin-ferrocene dyads. Also, the fluorescence of porphyrin was significantly quenched in porphyrin-ferrocene conjugates due to photoinduced electron transfer from ferrocene to porphyrin
- fluorescence investigation of hybrid 96 revealed that the fluorescence of zinc porphyrin was strongly quenched, and the electron-transfer quenching mechanism was validated by easy oxidation. Furthermore, femtosecond transient-absorption spectrum investigations offered proof of the charge-separation process









































NaI/PPh3-catalyzed visible-light-mediated decarboxylative radical cascade cyclization of N-arylacrylamides for the efficient synthesis of quaternary oxindoles
Beilstein J. Org. Chem. 2023, 19, 57–65, doi:10.3762/bjoc.19.5

- run in a similar fashion to related well-documented previous reports [54][68][69][70][71][72][73][74][75][76][77], through a light-induced, phosphine-assisted, intermolecular electron transfer from sodium iodide to the redox-active ester. Conclusion In summary, we developed an effective photocatalytic






Combining the best of both worlds: radical-based divergent total synthesis
Beilstein J. Org. Chem. 2023, 19, 1–26, doi:10.3762/bjoc.19.1

- led to common scaffolds 47 and diene 48 after subsequent elimination. Those molecules serve as templates for Ni-based radical-based sp3–sp2 coupling and single-electron transfer (SET)-based [3 + 2] coupling, respectively (Scheme 4). Initial attempts to realize the [3 + 2] radical coupling with CAN led
- radical to circumvent the unsuccessful Friedel–Crafts reaction. Prior reports implicated β-keto radical formation in the ring opening of siloxycyclopropanes with photoinduced electron transfer (PET) to 1,4-dicyanonaphthalene [65]. Inspired by reports on dual photoredox and Ni-catalytic cross-coupling





















Redox-active molecules as organocatalysts for selective oxidative transformations – an unperceived organocatalysis field
Beilstein J. Org. Chem. 2022, 18, 1672–1695, doi:10.3762/bjoc.18.179

- unfavorable (DABCO, quinuclidine) and in the case of triarylamines containing no hydrogen atoms at carbon atoms connected to the cation radical center. Cation radicals of aromatic amines or N-heterocyclic cation radicals are usually involved in electron transfer processes due to their low affinity to hydrogen
- mainly as mediators of oxidative electron transfer (SET mechanism). For example, one-electron oxidative properties of triarylamine cation radicals were used for the vinylarene difunctionalization with the formation of thiadiazolidine 1,1-dioxides that can be further transformed to the corresponding 1,2
- via a SET mechanism. The first electron transfer occurs from the aromatic system to the organocatalyst cation radical. After the deprotonation of the benzylic position a benzylic radical is formed. Then, a benzylic cation is produced by a second oxidative electron transfer followed by nucleophile








































Molecular and macromolecular electrochemistry: synthesis, mechanism, and redox properties
Beilstein J. Org. Chem. 2022, 18, 1505–1506, doi:10.3762/bjoc.18.158

- /bjoc.18.158 Keywords: electron transfer; electrosynthesis; organic electrochemistry; redox-active materials; Electrochemistry is now a powerful tool in organic chemistry not only for analyzing the electron transfer behavior of organic molecules and macromolecules, but also for driving organic
- , polymer electrolyte membrane electrolysis technology, and new methods coupled with photoredox catalysts or transition metal catalysis, resulting in remarkable progress in organic electrosynthetic processes. Theoretical calculations have also led to a better understanding of the electron transfer behavior
- batteries. By understanding organic electron transfer reactions, we can face the challenge of how to design materials with better cycle properties by suppressing undesired side reactions. To showcase this area of research, the present thematic issue focuses on the recent advances in molecular and
Cytochrome P450 monooxygenase-mediated tailoring of triterpenoids and steroids in plants
Beilstein J. Org. Chem. 2022, 18, 1289–1310, doi:10.3762/bjoc.18.135

- electron transfer from a reductase partner protein (step 2). The most common electron donors in plants are cytochrome P450 reductases which employ NADPH for the electron transfer, but several other electron transfer systems are known [22]. The reduced ferrous intermediate C, bearing an overall negative







Thermally activated delayed fluorescence (TADF) emitters: sensing and boosting spin-flipping by aggregation
Beilstein J. Org. Chem. 2022, 18, 1177–1187, doi:10.3762/bjoc.18.122

- developed mainly by focusing on photophysical processes, such as photoinduced electron transfer (PET), excited-state intramolecular proton transfer (ESIPT), fluorescence resonance energy transfer (FRET), etc. [21][22][24][25]. Several literature reports have also demonstrated the switching of fluorescence









Radical cation Diels–Alder reactions of arylidene cycloalkanes
Beilstein J. Org. Chem. 2022, 18, 1100–1106, doi:10.3762/bjoc.18.112

- to be much more effective in the reaction than other cycloalkanes. Since the reaction is completed with a substoichiometric amount of electricity, a radical cation chain pathway is likely to be involved. Keywords: arylidene cycloalkane; Diels–Alder reaction; radical cation; single-electron transfer
- ; spiro ring system; Introduction Single-electron transfer is one of the simplest modes for small molecule activation, employing a polarity inversion to generate radical ions which have proven to be unique reactive intermediates in the field of synthetic organic chemistry. A radical cation Diels–Alder
- reaction is a typical example of this activation mode since both the original diene and dienophile are electron-rich and thus not an effective combination of reactants [1][2][3][4][5][6][7][8][9][10][11]. Single-electron transfer makes the construction of six-membered ring systems possible. In general








Electrochemical formal homocoupling of sec-alcohols
Beilstein J. Org. Chem. 2022, 18, 1062–1069, doi:10.3762/bjoc.18.108

- from the corresponding vic-1,2-diol. Water may play a role as a proton source to facilitate the formation of the protonated ketyl radical through a concerted proton-electron transfer toward the ketone or smooth protonation of the radical anion species, which readily dimerize to vic-1,2-diol 2a [46][49







Cathodic generation of reactive (phenylthio)difluoromethyl species and its reactions: mechanistic aspects and synthetic applications
Beilstein J. Org. Chem. 2022, 18, 872–880, doi:10.3762/bjoc.18.88

- place. Thus, it was found that o-phthalonitrile should work as an electron transfer catalyst, i.e., a redox mediator. On the bases of the cyclic voltammetric measurements, the cathodic reduction of 1 was carried out at a constant potential using o-phthalonitrile as mediator. As shown in Scheme 5, the
- one- and two-electron transfer, their indirect cathodic reduction using mediators undergoes one-electron reduction selectively as reported by Saveant et al. [24]. In this study, we also confirmed that the o-phthalonitrile-mediated reduction of PhSCF2Br (1) in the absence of radical trapping reagents













Complementarity of solution and solid state mechanochemical reaction conditions demonstrated by 1,2-debromination of tricyclic imides
Beilstein J. Org. Chem. 2022, 18, 746–753, doi:10.3762/bjoc.18.75

- also found in the reaction of bicyclo[2.2.2] dibromide 42 (Scheme 3), which suggests that the mechanism may involve radical anion intermediates. This result further supports a single electron transfer (SET) and radical anion mechanism which was postulated earlier for the Zn/Ag debromination reaction







Structural basis for endoperoxide-forming oxygenases
Beilstein J. Org. Chem. 2022, 18, 707–721, doi:10.3762/bjoc.18.71

- the water addition occurs on the C3' carbocation, which is generated by one electron transfer to the ferric iron from the C3' radical intermediate (carbocation mechanism in Scheme 9). Notably, in this last step, the stereochemistry of the hydroxylation reaction is regulated by the enzyme to be the R














Direct C–H amination reactions of arenes with N-hydroxyphthalimides catalyzed by cuprous bromide
Beilstein J. Org. Chem. 2022, 18, 647–652, doi:10.3762/bjoc.18.65

- -electron transfer (SET) between CuBr and intermediate 5 forms intermediate 6, which initiates the N–O bond homolytic cleavage resulting in forming an N-centred phthalimidyl radical 7 (PhthN•) and anion 8. Meanwhile, Cu(I) is oxidized to Cu(II) in this step. Next, radical 7 attacks the benzene via radical




